
















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Raynaud-Syndrom: Altbekanntes und Neues zur Therapie
11. Januar 2013
Unter Anwendung strenger Kriterien (kalte Finger + Taubheitsgefühl + Zwei-Farben-Reaktion) liegt die Prävalenz des Raynaud-Syndroms (RS) bei Frauen um 2,9% und bei Männern um 0,5%1. Aufgrund der unterschiedlichen Prognose sowie der notwendigen weiteren Abklärung muss zwischen primärem und sekundärem RS differenziert werden: Ersteres wird als rein funktionelle Störung der die Akren versorgenden Blutgefäße ohne Grunderkrankung gesehen. Zweiteres tritt im Rahmen einer systemischen Erkrankung auf.
Klinische Manifestation und Diagnostik
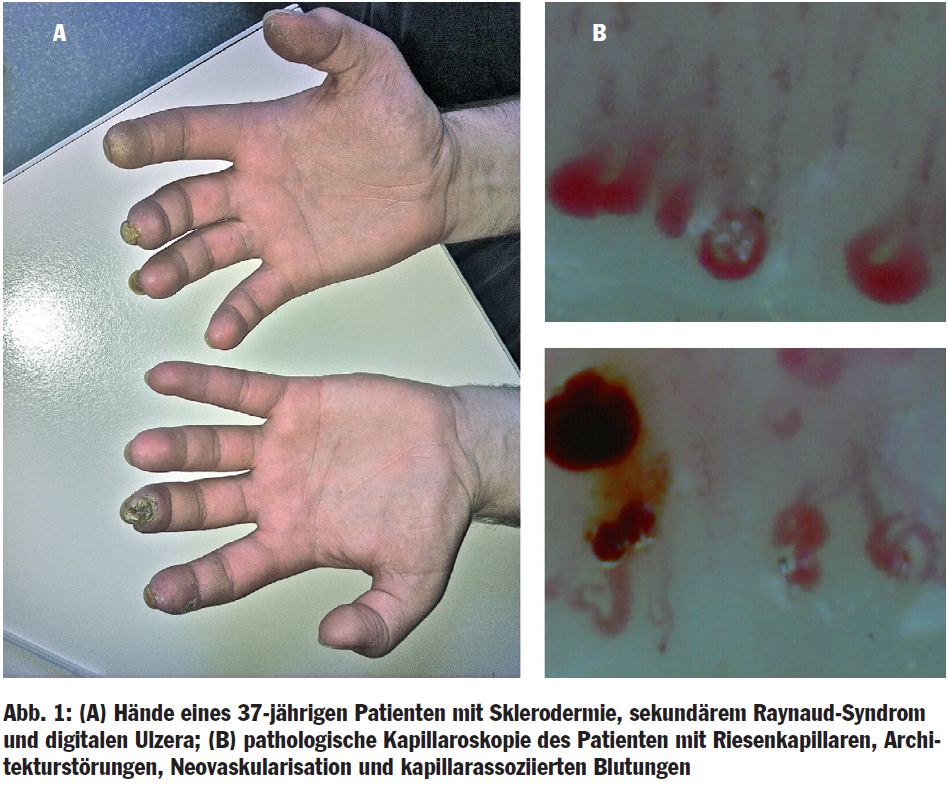
Das primäre RS manifestiert sich klassischerweise symmetrisch an beiden Händen (bzw. Vorfüßen) unter Aussparung der Daumen. Es treten meist keine Ulzerationen auf. Die klinische Untersuchung, Kapillaroskopie, Laborparameter sowie die Immunserologie sind unauffällig. In einer prospektiven Studie entwickelte sich bei lediglich 2% der Patienten mit RS eine rheumatische Erkrankung, wenn initial die Immunserologie unauffällig war. Hinweise für ein sekundäres RS sind höheres Alter bei Erstmanifestation (> 30 Jahre), intensive schmerzhafte Episoden, asymmetrischer Befall von Händen oder Vorfüßen, Nachweis von Autoantikörpern, pathologische Kapillaroskopie (Abb. 1) und klinische Zeichen einer Kollagenose. Häufiger kommt es zu Störungen des Nagelwachstums, Ulzerationen, Nekrosen und Knochenatrophie. In der Differenzialdiagnostik des sekundären RS sollte an zahlreiche sehr unterschiedliche Erkrankungen und Medikamente/Toxine gedacht werden (Tab.).

Therapie
Die Behandlung des RS und insbesondere seiner ischämischen Läsionen ist komplex (Abb. 2), zeigt jedoch aufgrund rezenter Einsichten in die Pathogenese und durch die Identifizierung zentraler Mediatoren einen deutlichen Progress. Eine medikamentöse Therapie ist bei Patienten indiziert, die auf Allgemeinmaßnahmen (Vermeidung von Kälte, Stress, Nikotin, lokale Wärmeanwendungen etc.) nicht ausreichend ansprechen. Bei Auftreten digitaler Ulzera muss auch eine adäquate Lokaltherapie (Wundmanagement, Dèbridement) sowie bei Infektion eine Antibiose erfolgen.
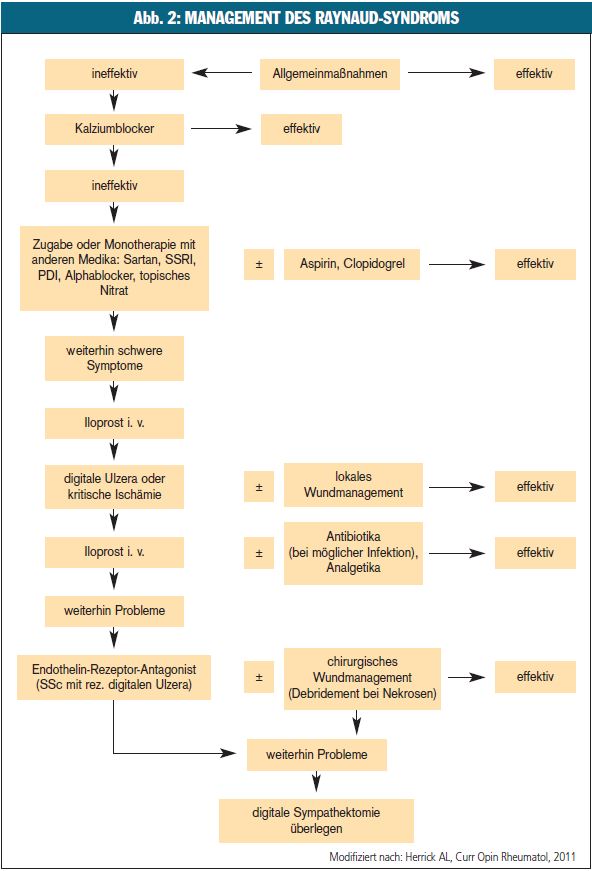
Kalziumantagonisten (KA, Nifedipin, Amlodipin) gelten als Medikamente der ersten Wahl zur Therapie des RS2. Rezente Studien belegen auch positive Effekte der KA auf die kardiale Beteiligung im Rahmen einer Sklerodermie (SSc). Bei unzureichender Effektivität können andere vasoaktive Medikamente zu KA kombiniert werden oder den KA ersetzen.
Angiotensinrezeptorblocker (Sartane) reduzieren Anzahl und Schwere der Raynaud-Attacken bei Patienten mit primärem und sekundärem RS3, während ACE-Hemmer in einer Studie keine positiven Effekte zeigten.
Selektive Serotoninwiederaufnahmehemmer (SSRI) zeigten positive Effekte in einer Studie und können vor allem bei Patienten mit ausgeprägten Nebenwirkungen auf die oben genannten Medikamente verwendet werden.
Prostazyklinanaloga: Bei kritischer Ischämie und digitalen Ulzerationen, aber auch bei schwerem RS ist intravenös verabreichtes Iloprost als Therapie der ersten Wahl anzusehen, wobei weder zur Dosierung noch zur Dauer dieser Therapie einheitliche Empfehlungen vorliegen2. An unserer Abteilung wird Iloprost in der Dosierung von 2 ng/min/kg (Dosistitration angezeigt) als kontinuierliche Infusion über sechs bis 24 Stunden verabreicht. Die Dauer der Verabreichung hängt vom Schweregrad des RS bzw. der Ausprägung digitaler Ulzera ab und beträgt im Durchschnitt sieben bis zehn Tage. Auch eine ambulante Verabreichung einmal pro Monat, zusätzlich zu anderen Therapien, hat sich im klinischen Alltag bewährt.
Endothelinrezeptorantagonisten: Endothelin ist ein starker Vasokonstriktor in systemischen und pulmonalen Blutgefäßen und hat zusätzliche Effekte auf das vaskuläre Remodeling und die Fibrose. Endothelin-1 gilt als ein zentrales Enzym in der Pathogenese der SSc. Endothelinrezeptorantagonisten (Bosentan) verbessern die Symptomatik des RS nicht, werden jedoch bei rezidivierenden digitalen Ulzera im Rahmen der SSc eingesetzt und reduzieren die Zahl der neu auftretenden Ulzera4, 5. Kombinationen mit Iloprost oder anderen vasoaktiven Substanzen sind möglich und bei einzelnen Patienten immer wieder notwendig. Phosphodiesteraseinhibitoren (PDI) (Sildenafil, Vardenafil, Tadalafil) zeigten in klinischen Studien überwiegend positive Ergebnisse, insbesondere auch bei sekundärem RS. Stickstoffmonoxid (NO) ist ein potenter Vasodilatator. PDI steigern den Effekt von NO über die Verminderung des Abbaus von zyklischem Guanosinmonophosphat. Der Blutfluss in den Akren wird verbessert, die Zahl und Schwere der Raynaud-Attacken reduziert. PDI wurden auch als Add-on-Therapie bei SSc eingesetzt und zeigten positive Auswirkungen auf digitale Ulzera6, 7. Obwohl PDI bereits häufig zur Therapie des RS und digitaler Ulzera eingesetzt werden, gelten sie noch nicht als ausreichend validierte Therapie2.
Thrombozytenfunktionshemmer: Patienten mit SSc zeigen verstärkte Thrombozytenaktivierung und -aggregation. Daher können Thrombozytenfunktionshemmer (Aspirin, Clopidogrel) bei SSc-assoziiertem schwerem RS und/oder kritischerIschämie eingesetzt werden2.
Statine könnten über mehrere Wege positiv auf das RS und begleitende ischämische Komplikationen wirken. Atorvastatin reduzierte in einer Untersuchung die Anzahl der neuen Ulzera und die Schwere des RS8. Die vorliegenden Daten erlauben jedoch keine generelle Therapieempfehlung für Statine bei RS.
Weitere Therapieoptionen
Die Lokaltherapie des RS mit Nitraten zeigt dosisabhängig lokale wie auch systemische Effekte. Rezent wurden positive Effekte bei Verwendung eines Nitroglyzerin-Patches beschrieben. Medikamente, die in kleinen Fallserien positive Effekte zeigten, sind Antioxidantien (Azetylzystein) und Botulinumtoxin. Für weitere Medikamente laufen zurzeit bereits klinische Studien. Untersucht werden unter anderem Treprostinil, verschiedene PDI, Endothelinrezeptorantagonisten und ein RhoA/Rho-Kinase-Inhibitor.
Literatur:
1 Bartelink MI et al., Neth J Med 1992
2 Herrick AL, Curr Opin Rheumatol 2011
3 Herrick AL, Curr Opin Rheumatol 2011
4 Nguyen VA et al., Rheumatology 2010
5 Matucci-Cerinic M et al., Ann Rheum Dis 2011
6 Shenoy PD et al., Rheumatology 2010
7 Brueckner CS et al., Ann Rheum Dis 2010
8 Abou-Raya A et al., J Rheumatol 2008

Ursprünglich erschienen:
AEK 01|2013
AEK 01|2013