
















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
MCTD oder Mischkollagenose – Paradigmenwechsel hinsichtlich Zuordnung und Diagnosekriterien
23. Juli 2012
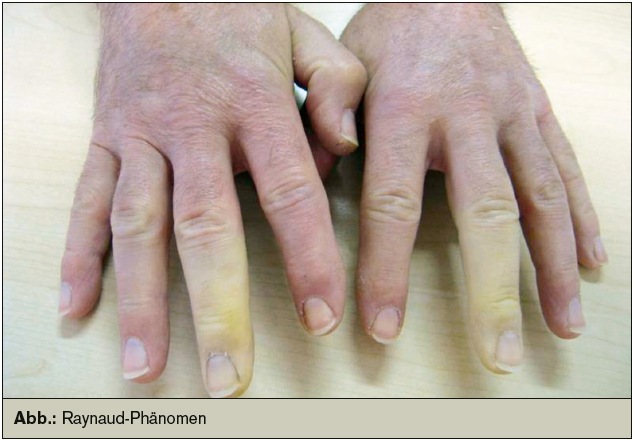 Ursprünglich schrieben Sharp und Kollegen der Mixed Connective Tissue Disease (MCTD) eine gute Prognose zu; nicht zuletzt aufgrund zunächst scheinbar fehlender Manifestation an inneren Organen und wegen des guten Ansprechens auf niedrig dosiertes Kortison. Bald allerdings mussten die Erstbeschreiber dieser neuen Kollagenose ihr ursprüngliches Konzept überarbeiten. Bei Beobachtung des Krankheitsverlaufs stellte sich heraus, dass sowohl innere Organe in Mitleidenschaft gezogen werden können und dass nicht jeder Betroffene von der alleinigen Glukokortikoidtherapie profitiert. Viele Symptome, deren Kombination ursprünglich als MCTD wegweisend beschrieben wurden, werden immer noch, obwohl unspezifisch, als MCTD-charakteristisch angesehen; so beispielsweise das Raynaud-Phänomen, „puffy hands“, Polyarthralgien oder Polyarthritis und Muskelschwäche. Diese Symptome sind besonders in der Initialphase der Erkrankung stark ausgeprägt. Im Verlauf treten Organmanifestationen und weitere Erscheinungen, wie sie für den systemischen Lupus erythematodes (SLE) oder die systemische Sklerose typisch sind, hinzu.
Ursprünglich schrieben Sharp und Kollegen der Mixed Connective Tissue Disease (MCTD) eine gute Prognose zu; nicht zuletzt aufgrund zunächst scheinbar fehlender Manifestation an inneren Organen und wegen des guten Ansprechens auf niedrig dosiertes Kortison. Bald allerdings mussten die Erstbeschreiber dieser neuen Kollagenose ihr ursprüngliches Konzept überarbeiten. Bei Beobachtung des Krankheitsverlaufs stellte sich heraus, dass sowohl innere Organe in Mitleidenschaft gezogen werden können und dass nicht jeder Betroffene von der alleinigen Glukokortikoidtherapie profitiert. Viele Symptome, deren Kombination ursprünglich als MCTD wegweisend beschrieben wurden, werden immer noch, obwohl unspezifisch, als MCTD-charakteristisch angesehen; so beispielsweise das Raynaud-Phänomen, „puffy hands“, Polyarthralgien oder Polyarthritis und Muskelschwäche. Diese Symptome sind besonders in der Initialphase der Erkrankung stark ausgeprägt. Im Verlauf treten Organmanifestationen und weitere Erscheinungen, wie sie für den systemischen Lupus erythematodes (SLE) oder die systemische Sklerose typisch sind, hinzu.
Langzeitstudien konnten zeigen, dass annähernd 30 % aller Patienten, bei denen mittels Klassifikationskriterien die Diagnose MCTD gestellt wurde, nach durchschnittlich 8 Jahren einen SLE, eine systemische Sklerose oder eine rheumatoide Arthritis entwickelten. Dies zusammen mit der Tatsache, dass das Auftreten der Anti-U1RNP-Antikörper (Ak) längst nicht so hochspezifisch ist wie ursprünglich angenommen, waren dafür ausschlaggebend, dass in den letzten 40 Jahren die MCTD als eigenständige Entität immer wieder stark kritisiert wurde.
Klinik
Initial stehen häufig Raynaud-Phänomen, „puffy hands“, Sklerodaktylie, Muskelschwäche oder Polyarthralgien im Vordergrund. Auch eine unspezifische Allgemeinsymptomatik wie Fieber, Müdigkeit und Gewichtsverlust sind häufig zu beobachten. Die Gelenkbeteiligung ist häufig; die Ausprägung sehr variabel und reicht von minimalen Arthralgien über Arthritis der kleinen oder großen Gelenke bis hin zu Erosionen und Deformierungen. Die Muskulatur ist gewöhnlich im Sinne einer entzündlichen Myopathie beteiligt. An der Haut zeigen sich sklerodermieartige Veränderungen. Photosensibilität, Schleimhautulzerationen, Exanthem und Vaskulitiszeichen an der Haut als lupustypische Erscheinungen treten ebenfalls auf. Das Raynaud- Phänomen ist bei über 90 % aller Betroffenen präsent.
Häufig manifestiert sich die MCTD – gegensätzlich zu Sharps ursprünglichem Krankheitskonzept – an inneren Organen. Die Lunge ist hierbei mit bis zu 85 % das am häufigsten betroffene Organ. Ähnlich wie bei der systemischen Sklerose entwickelt sich zumeist eine Lungenfibrose. Die Entwicklung eines pulmonalarteriellen Hochdrucks (PAH) verschlechtert die Prognose entscheidend. Die Veränderungen an der Lunge sind anfangs zumeist asymptomatisch. Im weiteren Verlauf leiden die Betroffenen zunehmend an Dyspnoe. Häufigstes Symptom bei gastrointestinaler Beteiligung ist die Dysphagie als Folge der Ösophagusmotilitätsstörung. Weitere mögliche gastrointestinale Erscheinungen im Rahmen der MCTD sind Vaskulitis der Mesenterialgefäße, Malabsorption, Diarrhö und chronisch aktive Hepatitis.
Die Perikarditis stellt die häufigste kardiale Manifestation dar. Auch Myokarditis, Überleitungsstörungen und Mitralklappenprolaps werden in Verbindung mit MCTD gebracht. Eine Nierenbeteiligung im Rahmen der MCTD zeigt sich hauptsächlich in Form einer membranösen und mesangialen Glomerulonephritis und ist im Vergleich zum Lupus selten. Die am häufigsten beobachteten hämatologischen Symptome sind Leukopenie, Anämie, Hypergammaglobulinämie und positiver Coombs- Test. Trotz fehlender Spezifität dieser hämatologischen Erscheinungen korrelieren sie mit der Krankheitsaktivität. Bei so manchem MCTD-Patienten manifestiert sich die Erkrankung auch im Nervensystem. Hier dominieren die Trigeminusneuralgie, Polyneuropathien und neuropsychiatrische Symptome. Bei MCTDPatienten mit aseptischer Meningitis konnten große Mengen an Anti-U1RNP-Ak im Liquor entdeckt werden.
Antikörper
Die antinukleären Antikörper (ANA) sind als Suchtest für Kollagenosen hoch positiv. Autoantikörper gegen U1RNP – eine der Hauptkomponenten des Spleißosoms – sind ein Erforderniskriterium für die Diagnose MCTD.
Niedrige Anti-U1RNP-Ak-Titer kommen auch beim SLE vor; hierbei handelt es sich vorwiegend um Anti-U1RNP-Ak vom IgM-Isotypen. Gleichzeitig sind oft beim Lupus Anti-Sm-Ak (die ebenfalls zu den ANA zählen) zu finden. Die PatientInnen mit hochtitrigen Anti-U1RNPAk erfüllen oft nicht ausreichend viele Klassifikationskriterien für SLE, systemische Sklerose (SSc) oder Poly-/Dermatomyositis. Im Lauf der Zeit ergeben die Symptome dann die MCTD-Kriterien („Sharp-Syndrom“).
Die Bezeichnungen Mischkollagenose oder Overlap-Syndrom beziehen sich auf die gleichzeitige Erfüllung von zwei Kriteriensets (z. B. SLE und rheumatoide Arthritis, auch als Rhupus beschrieben). Undifferenzierte Kollagenosen („vulgo Autoimmunprädisposition“) sind Ansammlungen einzelner Symptome (z. B. Raynaud-Phänomen und hohe ANA-Werte).
Diagnose
Die Vielfältigkeit der klinischen Symptomatik und subklinischen Manifestationen erschwert die Diagnosefindung entscheidend. Differenzialdiagnostisch sind stets frühe Krankheitsphasen aller anderen Kollagenosen sowie undifferenzierte Kollagenosen (mit nur vereinzelten Symptomen und hohen ANA-Werten) zu berücksichtigen.
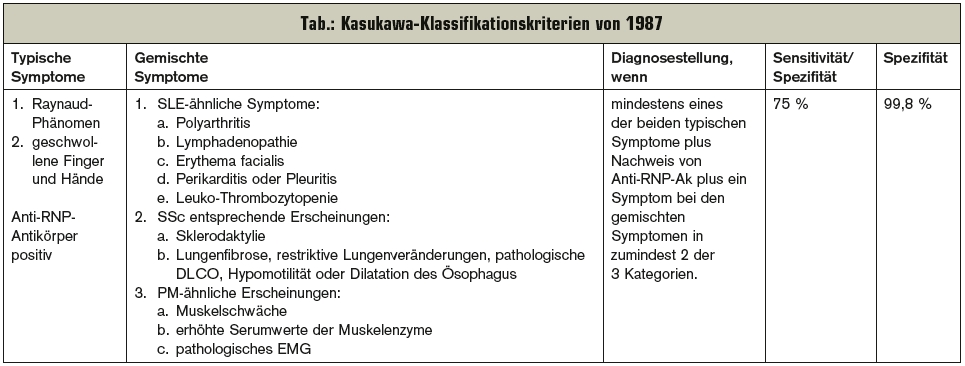
Zur Klassifikation der MCTD stehen unterschiedliche Kriterien zur Verfügung. In den letzten Jahren wurden mehrere Studien publiziert, die die Zuverlässigkeit, Sensibilität und Spezifität der zur Verfügung stehenden klinischen Kriterien miteinander verglichen. Die Kasukawa-Kriterien haben zumeist am besten abgeschnitten (Tab.). In der Praxis erfolgt die Diagnosestellung zumeist über den Nachweis hochtitriger Anti-U1RNP-Ak und die Erfüllung von zumindest 3 klinischen Kriterien. In einer rezenten Analyse von 161 Patienten bestand die Diagnose nach durchschnittlich 8 Jahren bei 58 % der Betroffenen weiterhin als MCTD, der Rest erfüllte mittlerweile andere Diagnosekriterien. Dies wird als Berechtigung für das Aufrechterhalten dieser diagnostischen Entität gesehen (Semin Arthritis Rheum 2012 Feb; 41 [4]:589-98.).
Therapie
Die MCTD hatte anfänglich den Ruf einer relativ harmlosen Bindegewebserkrankung mit guter Prognose. Ein extrem gutes Ansprechen auf die Therapie mit Glukokortikoiden wurde dieser Kollagenose zugeschrieben. Langzeitstudien widerlegten dies. So entwickelt ein Großteil der Betroffenen Organbeteiligungen mit zum Teil lebensbedrohlichen Manifestationen (s. o.). Auch stellte sich heraus, dass gerade die prognoseentscheidenden Manifestationen zumeist nicht mittels alleiniger Glukokortikoidtherapie beherrschbar sind. Nur ein Bruchteil der Patienten mit MCTD profitiert von einem leichten, selbstlimitierenden Krankheitsverlauf.
Da bezüglich der Therapie der MCTD kontrollierte klinische Studien fehlen, ist man angehalten, auf die konventionelle Therapie zurückzugreifen, die für die ähnlichen Manifestationen im Rahmen anderer Kollagenosen angewendet werden. In jedem Fall ist eine auf den Patienten mit seiner Symptomatik sowie der Organmanifestationen angepasste Therapie unerlässlich. Grundsätzlich sprechen entzündliche Prozesse wie Fieber, Serositis, Myositis, Arthritis sowie die für den Lupus charakteristischen Haut- und Schleimhautveränderungen gut auf Steroide an. Jedoch sollten hochdosierte Glukokortikoide zur Vermeidung seltener renaler Krisen nach Steroidgabe nur in Abwägung des Nutzen-Risiko- Profils eingesetzt werden. Sklerodermie-typische Erscheinungen wie Sklerodaktylie, ösophageale Dysfunktion, Raynaud-Phänomen und interstitielle Lungenkrankheit bedürfen in der Regel zytotoxischer Immunsuppressiva. Die am häufigsten verwendeten Immunsuppressiva sind Glukokortikoide (Prednisone und Methylprednisolon) sowie zytotoxisch wirkende Substanzen (zumeist Cyclophosphamid). Hydroxychloroquin, Methotrexat (MTX) und verschiedene vasodilatorisch wirksame Pharmaka werden ebenfalls, allerdings mit wechselndem Erfolg, zur Behandlung der MCTD eingesetzt. Um lebensbedrohlichen Komplikationen bestmöglich vorzubeugen, sind die regelmäßige Überprüfung der Lungenfunktion und -morphologie sowie des PAH mittels CO-Diffusionsmessung (DLCO), Computertomografie des Thorax und Belastungsechokardiografie unerlässlich.
FACT-BOX
• Zu den häufigsten Manifestationen der MCTD zählen das Raynaud-Phänomen, Arthralgien, Ösophagusmotilitätsstörungen, Wurstfinger, „puffy hands“ und Muskelschwäche.
• Hohe Anti-U1rnP-Ak-Titer in Abwesenheit von Anti-Sm-Ak sind kennzeichnend für MCTD.
• Ein individualisiertes Behandlungsschema je nach Symptomatik und Beteiligung innerer Organe unter Verwendung der konventionellen Therapeutika für die gleichen Manifestationen im Rahmen anderer Kollagenosen ist notwendig.

Ursprünglich erschienen:
UIM 04|2012
UIM 04|2012
Herausgeber: Univ.-Prof. Dr. Günter J. Kreijs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2012-05-25
Zur Ausgabe »
Publikationsdatum: 2012-05-25
Zur Ausgabe »