

























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur RegistrierungVersuch einer Typologie der Myelomerkrankung
Mein Zugang zur Erstlinientherapie des Myeloms
6. Juli 2023
Ein Disclaimer zur Einleitung:
1) Aufgrund des essayistischen Charakters dieses Beitrages erlaubt sich der Autor, Behandlungsergebnisse verschiedener Studien direkt miteinander zu vergleichen („cross-trial comparisons“).
2) Aus demselben Grund werden in diesem Beitrag persönliche Betrachtungen und Einschätzungen vorgenommen.
3) Explizit ausgenommen ist in diesem Beitrag die Diskussion patientenbezogener Faktoren, die für die Therapieplanung herangezogen werden müssen (Frailty, Komorbidität u. a.), was die Bedeutung ebendieser Faktoren keinesfalls in Frage stellen soll.
Wo stehen wir heute? Was sagen uns heutige Studien über morgen?
Ohne Zweifel haben die Fortschritte in der medikamentösen Therapie des Myeloms den Charakter und Verlauf dieser Erkrankung grundlegend geändert. Beispielhaft sei eine retrospektive Analyse des University Hospital of Salamanca erwähnt1, in der Patient:innen mit Diagnosestellung von 1980 bis 1990 ein Gesamtüberleben von 22,4 Monaten aufwiesen, Patient:innen der rezentesten Kohorte mit Diagnose 2011 bis 2020 ein Gesamtüberleben von 103,6 Monaten. Patient:innen mit t(11;14) etwa dürften aber von modernen Kombinationsregimen (verglichen mit konventioneller Chemotherapie) nicht in gleicher Weise profitiert haben wie andere Patient:innen der Standardrisikogruppe oder auch die der Hochrisikogruppe.2 Ursache hierfür dürfte die gute Empfindlichkeit der durch t(11;14) charakterisierten Myelome gegenüber konventioneller Chemotherapie sein (neben besonderer Sensitivität – wie späterhin festgestellt – gegenüber dem Bcl-2-Inhibitor Venetoclax). Traditionell galt daher die t(11;14) als Indikator für eine eher günstige Prognose.3 Heute stellt sich dieses Bild durch den Einfluss zytogenetischer Kofaktoren differenzierter dar. In diesem Zusammenhang seien eigene Daten einer retrospektiven Analyse erwähnt, die einen ungünstigen Einfluss eines Zugewinnes von 1q (1q+) bei jüngeren, fitten, mit Hochdosistherapie und autologer Stammzelltransplantation behandelten Patient:innen mit durch t(11;14) charakterisiertem Myelom belegen.4
Leider fehlen uns heute noch gute prädiktive Marker, die eine verlässliche Stratifizierung von Myelompatient:innen für die Erstlinientherapie erlauben würden. Aktuell stehen Protokolle sehr unterschiedlicher Therapieintensität zur Verfügung, wobei der Einsatz einer Hochdosistherapie mit autologer Transplantation vorwiegend an Alter und Fitness der Patient:innen festgemacht wird. Durch neue immunologische Therapieverfahren, etwa „CAR T-cells“ und/oder bispezifische Antikörper, die aktuell in klinischen Studien auch in der Erstlinientherapie erprobt werden, dürfte wohl in einigen Jahren die Hochdosistherapie in den Hintergrund gedrängt, wenn nicht sogar obsolet werden. Mit Daratumumab + Lenalidomid/Dexamethason (DRd) können ohne intensivierte Konsolidierung hervorragende Überlebensdaten erzielt werden5, das mediane progressionsfreie Überleben (PFS) liegt vermutlich (bis jetzt „not reached“) bei über 5 Jahren. Mit dem zusätzlichen Einsatz von etwa bispezifischen Antikörpern in der ersten Linie (z. B. aktuell Teclistamab in der MajesTEC-7-Studie geprüft) wird eine Verlängerung des medianen PFS auf etwa 7 Jahre antizipiert. Mit autologer Transplantation und einem modernen Induktionsregime (allerdings noch ohne Inklusion eines Anti-CD38-Antikörpers) lag das mediane PFS in der EMN02/HO95-Studie bei 56,7 Monaten6, in der IFM/DFCI-2009-Studie bei 50 Monaten7, im DETERMINATION-Trial bei 67,5 Monaten7a, also unter 6 Jahren. Auch wenn sich mit Inklusion eines Antikörpers wie Daratumumab in die Induktionstherapie weitere Verbesserungen des PFS auch in der intensiven Erstlinienschiene verzeichnen lassen (4-Jahres-PFS in der GRIFFIN-Studie 87,2 %8, oder siehe auch CASSIOPEIA-Studie9), besteht doch der deutliche Eindruck, dass mit den moderneren immuntherapeutischen Verfahren der bis dato belegbare Unterschied zwischen Hochdosis ja/nein in der Konsolidierungstherapie zum Schwinden gebracht wird.
Da durch die erfolgreichen Therapieverfahren längere und zunehmend auch sehr lange Krankheitsverläufe beobachtet werden, müssen mögliche Spättoxizitäten, wie vor allem hämatologische und solide Sekundärneoplasien nach Gabe von Melphalan in den Fokus gerückt werden (bekannterweise wird diese Problematik durch eine Lenalidomid-Erhaltungstherapie noch aggraviert). Im Myeloma-XI-Trial waren bei 12,2 % der Patient:innen nach Transplantation und mit Lenalidomid-Maintenance-Therapie Sekundärneoplasien zu verzeichnen.10
Zytogenetische Klassifikation im Umbruch
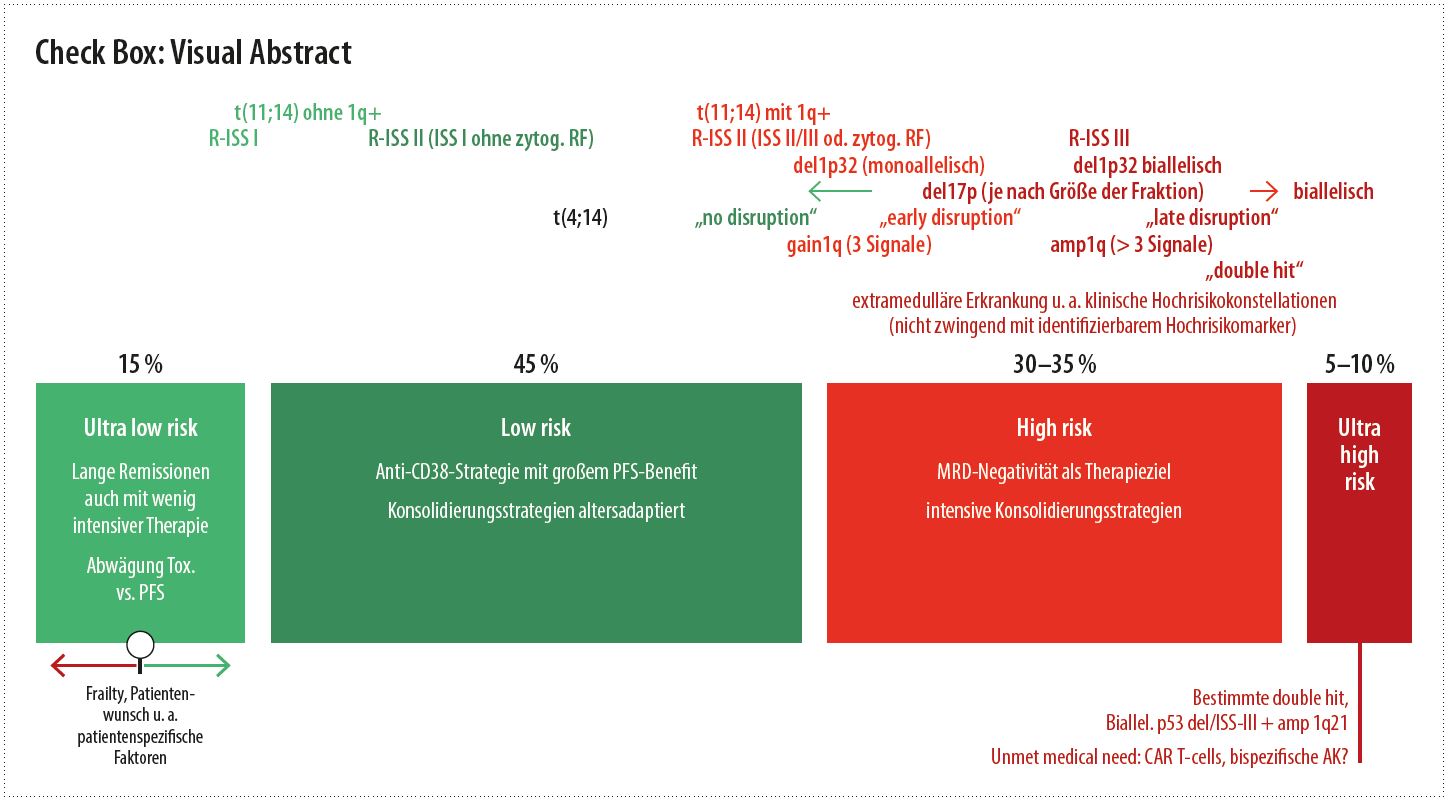
Eine gewisse Hilfe für eine Therapiestratifizierung bieten die klinischen und zytogenetischen Risikofaktoren, allerdings mit Limitationen. Es ist bekannt, dass im R-ISS-II eine sehr heterogene Prognosegruppe zusammengefasst ist. Patient:innen mit alleiniger Erhöhung der Lactatdehydrogenase (LDH) mit ISS I und ohne zytogenetische Risikofaktoren haben eine deutlich bessere Prognose, als wenn ISS II bzw. III oder zytogenetische Faktoren für die R-ISS-Klasse-II-Zuteilung verantwortlich sind.11 Das European Myeloma Network (EMN) hat bereits einen Vorschlag zur Revision der Klassifikation vorgelegt (R2-ISS)12, mit Inklusion des zytogenetischen Risikofaktors 1q+ zur Erstellung von 4 Risikogruppen („low“, „low-intermediate“, „intermediate-high“, „high“). 1q+ („gain“ = 3 Signale, „amplification“ = 4 oder mehr Signale) stellt die häufigste Risikoaberration beim Myelom dar13 und ist bei etwa 40 % der Patient:innen schon bei Diagnosestellung nachweisbar (70 % im Relaps; zur Häufigkeit der in diesem Beitrag angesprochenen Aberrationen siehe auch Tabelle). Weitere Erkenntnisse zu den zytogenetischen Risikofaktoren, die vermutlich in neue, adaptierte Versionen des R-ISS einfließen werden, betreffen die Charakterisierung der 1p32-Deletion (11 % der Fälle; monoallelisch oder biallelisch) als Risikofaktor14, die Feststellung, dass die klonale Fraktion für die Bedeutung der 17p-Deletion als Risikofaktor eine Rolle spielt15, und die Charakterisierung von verschiedenen Bruchpunkten bei t(4;14) mit unterschiedlicher prognostischer Signifikanz.16 Zu den letzteren beiden Punkten seien folgende Details angemerkt: Für die 17p-Deletion wurde eine Fraktion von > 55 % Myelomzellen mit Deletion (dies findet sich bei etwa 2/3 der Fälle mit 17p-Deletion) als idealer Cut-off für eine prognostische Signifikanz definiert.15 Kritisch sei angemerkt, dass die Überlebenskurven (del17p ≤ 55 % oder > 55 %) für das Gesamtüberleben eindrucksvoller zu separieren scheinen als für das PFS, sodass für letzteren Parameter möglicherweise doch auch kleinere Myelomzellfraktionen mit 17p-Deletion eine klinisch relevante prognostische Bedeutung haben könnten. Für die t(4;14) wurden 3 Bruchpunkte charakterisiert, mit Expression unterschiedlicher MMSET-(NSD2-)Fusionstranskripte16 und unterschiedlicher prognostischer Signifikanz („late disruption“ 31,3 % schlechte Prognose, „early disruption“ 23,7 % intermediäre Prognose, „no disruption“ 45 % bessere Prognose). Mittels FISH-Technik werden sich diese unterschiedlichen Bruchpunkte nicht charakterisieren lassen, sodass hier wohl Sequenzierungsverfahren in die Routinediagnostik Eingang finden müssten, um diese Differenzierung treffen zu können.
Obwohl der prognostische Wert der klinischen und zytogenetischen Risikofaktoren gut etabliert ist, ist Prädiktion (welche Therapie für welche Patient:innen) allerdings nur in sehr grobem Ausmaß möglich. So wissen wir etwa aus vorliegenden Studien (MAIA5, ALCYONE17, CASSIOPEIA9, auch GRIFFIN8), dass der Vorteil durch Inklusion von Daratumumab in die Erstlinieninduktionstherapie besonders bei Standardrisiko, weniger bei Hochrisikopatient:innen, zum Tragen kommt.

Typologie anhand klinischer Verläufe
Vielleicht kann es hilfreich sein, in umgekehrter Vorgehensweise klassische klinische Verläufe zu definieren, gewissermaßen eine Typologie des Myeloms zu erstellen (vielleicht in bewusster Reduktion auf Stereotypen), um dann zu diskutieren, welches therapeutische Gewand am besten zu dem jeweiligen Typus passen könnte.
Aus meiner Sicht lassen sich solcherhand 4 Grundformen der Myelomerkrankung abgrenzen, wobei jeweils 2 in den Standardrisiko- und 2 in den Hochrisikobereich fallen (zytogenetisch nach der Mayo-Clinic-Stratifizierung mSMART18 sind etwa 60 % der neudiagnostizierten Myelomerkrankungen als Standardrisiko, 40 % als Hochrisiko zu klassifizieren).
Standardrisikobereich
Im Standardrisikobereich würde ich eine „Ultra-low risk“-Gruppe heraus separieren wollen, bei der sich auch mit wenig intensiver Therapie ein langes PFS erzielen lässt. So lag etwa das 4-Jahres-PFS mit Lenalidomid + Dexamethason (Rd) in der FIRST-Studie19 bei Patient:innen, die zumindest eine sehr gute partielle Remission (VGPR) erreicht haben, bei 53,8 % und somit in einem Bereich, der in der EMN02/HO95-6 und IFM/DFCI-Studie7 mit wesentlich intensiverer Therapie erkauft wurde. Derzeit gibt es leider keinen guten Marker, um diese „Ultra-low risk“-Gruppe vorab aus der Standardrisikogruppe heraus zu separieren, wiewohl das aus meiner Sicht ein dringlich zu lösendes medizinisches Problem darstellt („unmet medical need“). Warum sollten diesen Patient:innen intensivere Therapieverfahren mit mehr Nebenwirkungen und langfristig immunsuppressiver Wirkung zugemutet werden, wenn dies auch mit weniger toxischer Therapie möglich ist? Selbst wenn in dieser Gruppe mit Therapieeskalation eine Verlängerung der Krankheitskontrolle möglich wäre, ist die Signifikanz bzw. klinische Wertigkeit dieses Vorteils mit der Toxizität und den Nebenwirkungen der zusätzlichen Therapie in Abwägung zu bringen, etwa mit dem Parameter der „quality-adjusted time without symptoms of disease and toxicity“ (Q-TWIST).20
Im zweiten Standardrisikobereich überwiegen die Vorteile einer eskalierten Induktionstherapie im Sinne der Gabe zumindest eines Triplets (heute wohl vorwiegend Dara-Rd analog MAIA; HR für PFS in der Standardrisikogruppe 0,49 vs. 0,85 in der Hochrisikogruppe5) oder Quadruplets (Dara-VRd analog GRIFFIN8). Hinsichtlich einer Konsolidierungstherapie wird wohl bei jüngeren Patient:innen dieser Gruppe eine Hochdosistherapie mit autologer Transplantation in erster Linie nach wie vor zu den Therapieempfehlungen zählen. Ich würde aber vermuten, dass dieses Vorgehen ein baldiges Ablaufdatum hat, das erreicht ist, sobald bispezifische Antikörper und/oder „CAR T-cells“ in erster Linie eingesetzt werden können (siehe auch Anmerkungen weiter oben).
Hochrisikobereich
In der ersten (hauptsächlichen, zahlenmäßig umfangreicheren) Hochrisikogruppe ist als wichtiges Therapieziel das Erreichen einer „Minimal residual disease“-(MRD-)Negativität zu definieren, da Patient:innen mit Hochrisikoerkrankung typischerweise schnell wieder relapsieren, wenn MRD nachweisbar bleibt.21 Bei Erreichen von MRD-Negativität kann die ungünstige Prognose in der Hochrisikogruppe soweit ausgeglichen werden, dass sich die Überlebenskurven derjenigen der Standardrisikogruppe angleichen.22 Somit sind eskalierte Konsolidierungsstrategien von Einfach- bis Doppeltransplantation bis zu Konsolidierungsprotokollen post autologe Stammzelltransplantation (ASCT), wie etwa in der ATLAS-Studie geprüft23, für diese Subgruppe wichtige Therapiebausteine, die vielleicht auch nicht ganz so leicht durch alternative immunologische Verfahren ersetzt werden können, wie in der Standardrisikogruppe zu erwarten ist. Das Erreichen von MRD-Negativität hat im Hochrisikobereich eine ungleich größere Bedeutung als im Standardrisikobereich, da im letzteren auch weniger tiefe Remissionen langanhaltend stabil bleiben können (prinzipiell ist auch im Standardrisikobereich das Erreichen von MRD-Negativität mit besseren Überlebensdaten assoziiert; die Abwägung, ob intensivere Maßnahmen zur Erreichung dieses Therapiezieles eingesetzt werden sollen, muss hier allerdings differenzierter erfolgen).
Neben der Therapieintensität mit dem Ziel Erreichen von MRD-Negativität, insbesondere auch von sogenannter „sustained“ MRD-Negativität (siehe dazu etwa FORTE-Trial24), dürfte auch der Therapiedauer in der Hochrisikogruppe eine besondere Bedeutung zukommen.
Unlängst konnte gezeigt werden, dass bestimmte zytogenetische Hochrisikoaberrationen, sofern sie singulär vorkommen, mit einem besonders günstigen Effekt einer Lenalidomid-Erhaltungstherapie assoziiert sind.25 Dies trifft etwa für die del1p, del17p und t(4;14) zu, nicht jedoch für 1q+. Bei Patient:innen mit 2 oder mehr Risikoaberrationen („double hit“) erzielt eine Lenalidomid-Erhaltungstherapie hingegen nur einen limitierten Benefit.
Das leitet über zur zweiten (kleineren) Hochrisikogruppe: Patient:innen mit Myelomerkrankung, die zwar initial eine Remission erreichen (mitunter auch MRD-Negativität), die jedoch nur sehr kurzfristig ist. In den folgenden Salvage-Therapien besteht dann typischerweise nur ein minimales bis kein Ansprechen und sehr schnell muss ein refraktärer Verlauf konstatiert werden. Für diese Patient:innen ist durch therapieintensive Verfahren kaum eine Prognoseverbesserung zu erzielen (siehe etwa die „Double hit“-Fälle im MASTER-trial26) und daher ein „medical need“ zu postulieren. Wie für die „Ultra low risk“-Gruppe festgestellt, glaube ich, dass auch die „Ultra high risk“-Gruppe heute verlässlich nur durch den klinischen Verlauf definiert werden kann, wenngleich für „ultra high risk“ mit genomischen Analysen bereits recht treffsichere Klassifizierungen vorab möglich scheinen. Will man „ultra high risk“ eng definieren, fallen vermutlich nicht alle „Double hit“-Fälle in diese Kategorie, sondern nur Fälle mit bestimmten „double hits“. So konnte etwa mittels „next generation sequencing“ (NGS) eine Gruppierung von Myelomfällen, charakterisiert durch entweder biallelische p53-Inaktivierung und/oder ISS-Stadium III mit amp 1q21 (6,1 %), mit ausgesprochen schlechter Prognose separiert werden (medianes PFS 15,4 Monate, medianes Gesamtüberleben 20,7 Monate).27 Eine gewisse Hoffnung kann darin bestehen, dass sich die neuen immunologischen Therapieverfahren wie bispezifische Antikörper oder „CAR T-cells“ für diese sonst primär refraktär verlaufenden Myelomfälle als Gamechanger erweisen. Wenn ein Studieneinschluss für solche Patient:innen nicht möglich sein sollte, wird wohl typischerweise eine Therapiemaximierung mit zweifacher Hochdosistherapie mit autologer Transplantation gefolgt von zeitlich extendierten Konsolidierungsstrategien mit Proteasom-Hemmer + immunmodulatorischer Substanz empfohlen werden, mit den intrinsischen Limitationen im aktuellen State of the Art (wie oben angesprochen).
Resümee
Am Horizont beginnt sich die Möglichkeit eines Profilings jeder einzelnen Myelomerkrankung abzuzeichnen, das sowohl die genetische Signatur der Erkrankung wie auch die Komposition des Microenvironments in der Analyse berücksichtigt. Erste interessante Vorstöße in diese Richtung wurden bereits unternommen und distinkte Immunsignaturen charakterisiert.28 Möglicherweise werden anhand dieser Merkmale Vorhersagen über die Wirksamkeit spezifischer medikamentöser Interventionen getroffen werden können und somit individualisierte Therapiepfade festgelegt.
Referenzen: (1) Puertas B et al., Cancers (Basel) 2023; 15(5):1558 (2) Puertas B et al., Blood Cancer J 2023; 13(1):40 (3) Fonseca R et al., Blood 2002; 99(10): 3735–41 (4) Udovica S et al., Impact of additional chromosomal aberrations in multiple myeloma with t(11;14). Focus on gain or amplification 1q21. OeGHO- Frühjahrstagung 2023; E-Poster H10 (5) Facon T et al., Lancet Oncol 2021; 22(11):1582–96 (6) Cavo M et al., Lancet Haematol; 7(6):e456–e468 (7) Attal M et al., NEJM 2017; 376(14):1311–20 (7a) Richardson P et al., NEJM 2022; 387(2):132–47 (8) Sborov DW et al., Presented at IMS 2022; August 25–27, 2022; Abstract OAB-057 (9) Moreau P et al., Lancet 2019; 394(10192):29–38 (10) Jones et al., Blood 2022; 140 (Suppl 1): 1823–25 (11) Schavgoulidze A et al., Haematologica 2022 Sep 29. Epub ahead of print (12) D’Agostino M et al., JCO 2022; 40(29): 3406–18 (13) Schmidt TM et al., Blood Cancer J 2021; 11(4):83 (14) Schavgoulidze A et al., Blood 2023; 141(11):1308–15 (15) Thakurta A et al., Blood 2019; 133(11):1217–21 (16) Stong N et al., Blood 2023; 141(13):1574–83 (17) Mateos MV et al., Lancet 2020; 395(10218):132–41 (18) Kumar SK et al., Mayo Clin Proc 2009; 84(12):1095–11 (19) Facon T et al., Blood 2018; 131(3):301–10 (20) Husson O and Jones RL, Cancer 2017; 123(12):2200–02 (21) Goicoechea I et al., Blood 2021; 137(1):49–60 (22) Paiva B et al., Blood 2016; 127(25):3165–74 (23) Dytfeld D et al., Lancet Oncol 2023; 24(2):139–50 (24) Gay F et al., Lancet Oncol 2021; 22(12):1705–20 (25) Panopoulou A et al., Blood 2023; 141(14):1666–74 (26) Costa LJ et al., JCO 2022; 40(25):2901–12 (27) Walker BA et al., Leukemia 2019; 33(1):159–70 (28) Larrayoz M et al., Nat Med 2023; 29(3):632–45

Autor:
OA Priv.-Doz. Dr. Niklas Zojer
OA Priv.-Doz. Dr. Niklas Zojer
1. Medizinische Abteilung
Zentrum für Onkologie und Hämatologie
Klinik Ottakring, Wien