


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Akromegalie
24. Mai 2019
Ursachen und Manifestation
Akromegalie ist auf eine gesteigerte unkontrollierte Produktion von Somatotropin (Wachstumshormon, GH) zurückzuführen. Ursache dafür ist nahezu immer ein GH-produzierendes Hypophysen-Adenom. Nur in Ausnahmefällen kann auch ein ektoper GH-Releasing-Hormon- produzierender Tumor verantwortlich sein. Die überschießende GH-Produktion verursacht neben den namensgebenden auch eine Vielzahl von weiteren Symptomen (Tab. 1).1

Die somatischen Veränderungen entwickeln sich dabei so langsam, dass die Betroffenen ebenso wie ihre nächste Umgebung diese oft jahrelang nicht bemerken. Somit vergehen durchschnittlich 7 Jahre vom retrospektiv eruierten ersten Auftreten von Symptomen bis zur Diagnose.2 Alle Bemühungen, das Intervall durch Erhöhung der Bekanntheit dieser seltenen Erkrankung zu verkürzen, sind bislang erfolglos geblieben. Dies ist auch deshalb so bedauerlich, weil zahlreiche der (die Lebensqualität der Betroffenen erheblich beeinträchtigenden) Begleiterkrankungen, wie z. B. Skelettveränderungen und das obstruktive Schlaf-Apnoe-Syndrom, auch bei Heilung der Akromegalie nicht oder nur geringfügig gebessert werden können. Unbehandelte Akromegalie ist mit einer auf das Doppelte erhöhten Mortalität und beträchtlicher Morbidität assoziiert.3
Diagnostik
Zur Diagnosestellung reicht eine einmalige Bestimmung von GH und des durch GH kontrollierten, vorwiegend in der Leber produzierten, Insulin-like Growth Factor-1 (IGF-1) meist nicht aus.3 Dies, obwohl IGF-1 eine deutlich längere Halbwertszeit als GH aufweist und weniger anderen äußeren Einflüssen wie etwa Stress unterworfen ist. Neben der Kontrolle der basalen GH- und IGF-1-Plasmakonzentration ist dazu meist ein Glukosesuppressionstest erforderlich. Wie beim oralen Glukosetoleranztest zur Diagnose von Störungen des Kohlenhydratstoffwechsels werden dabei 75 g Glukose oral verabreicht und GH basal und nach 30, 60, 90 und 120 Minuten gemessen. Zum Ausschluss des Vorliegens einer Akromegalie sollte GH dabei unter 0,4 ng/ml supprimiert werden. Durch die Bestimmung der Glukose-Konzentration basal und nach 2 Stunden kann gleichzeitig eine Störung des Kohlenhydrat-Stoffwechsels nachgewiesen bzw. ausgeschlossen werden.
An den laborchemischen Nachweis einer Akromegalie ist eine Magnetresonanztomografie (MRT) der Sella-Region anzuschließen. Dabei sollte neben der Größe des Adenoms auch seine Beziehung zum Chiasma opticum und dem Sinus cavernosus beschrieben werden. Bei Makroadenomen (Adenome mit einer Grösse über 1 cm) bzw. an das Chiasma opticum heranreichenden Adenomen sollte auch eine neuroophthalmologische Untersuchung zur Dokumentation etwaiger Gesichtsfeldausfälle angeschlossen werden. Weiters ist auch eine eventuell vorliegende Beeinträchtigung der übrigen Hypophysenvorderlappen-Funktionen zu überprüfen.3 Dazu ist die Bestimmung der basalen Plasma/Serum-Konzentrationen von fT4, T3, Testosteron bzw. Östradiol und Progesteron sowie Cortisol gut geeignet; bei niedrigen Cortisol-Werten sollte zusätzlich eine Messung der Cortisol-Ausscheidung im 24-Stunden-Harn erfolgen. Gegebenenfalls ist bereits präoperativ eine Substitutionstherapie mit Cortisol und/oder Thyroxin einzuleiten. Auch die Prolaktin-Serum-Konzentration sollte aufgrund der häufig vorliegenden Ko-Sekretion von Prolaktin mit GH kontrolliert werden.
Therapeutische Optionen
Bei Akromegalie stellt die transsphenoidale Entfernung des Adenoms die Therapie der ersten Wahl dar. Durch eine Operation können bis zu > 85 % der Patienten mit Mikroadenom geheilt werden, bei Makroadenomen sinkt dieser Anteil in Abhängigkeit von Größe und Lokalisation (Invasion des Sinus cavernosus) beträchtlich auf weniger als 50 %.4 Mögliche Nebenwirkungen der transsphenoidalen Operation sind eine Beeinträchtigung der übrigen Hypophysen-Funktionen (inklusive Diabetes insipidus), Liquorrhö, Meningitis und meist transiente Elektrolytstörungen. Jedenfalls sollte die Operation, wie auch die gesamte übrige Betreuung, Institutionen mit ausreichender Erfahrung auf dem Gebiet dieser seltenen Erkrankung vorbehalten bleiben.5
Wird durch eine Operation keine Heilung erzielt, stehen sehr effiziente medikamentöse Therapiealternativen zur Verfügung (Tab. 2). Dies gilt auch für den Fall, dass aufgrund von (meist kardialen, aber auch durch Veränderungen der Knorpel, Knochen und Schleimhaut im oberen Respirationstrakt bedingten) Komorbiditäten eine Operation bzw. Intubation nicht möglich ist oder dies vom Patienten oder der Patientin abgelehnt wird.
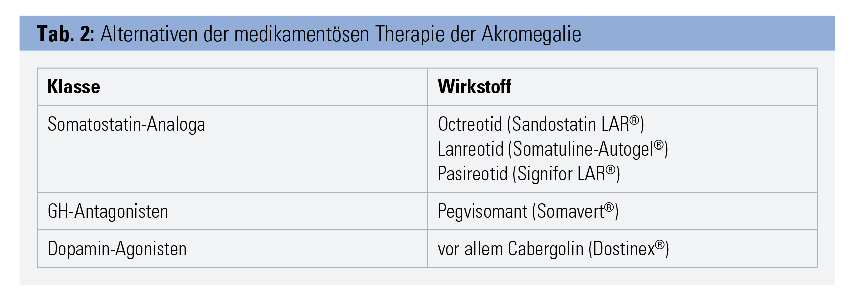
In erster Linie sind dabei die Somatostatin-(SMS-)Analoga der 1. Generation, Octreotid und Lanreotid, zu erwähnen. Beide sind in einer Galenik, die eine Applikation alle 4 Wochen erlaubt, verfügbar. Diese Therapie senkt nicht nur GH- und IGF-1-Plasmakonzentrationen, sondern führt auch meist zu einer Größenreduktion des Adenoms bzw. Tumorrests nach einer stattgehabten Operation. Nebenwirkungen dieser Therapien sind in Tabelle 3 angeführt. Bei nur geringer Aktivität der Akromegalie (IGF-1-Konzentration bis 50 % über der Obergrenze des Referenzbereichs) kann auch die Therapie mit dem Dopamin-Agonisten Cabergolin versucht werden. Falls mit SMS-Analoga der 1. Generation oder Cabergolin nicht das Therapieziel einer Normalisierung von IGF-1 und GH erzielt werden kann, kann eine Kombination dieser beiden Arzneiklassen zum Ziel führen. Als Therapie der 2. Wahl sind der GH-Antagonist Pegvisomant und das SMS-Analogon der 2. Generation, Pasireotid, verfügbar.6 Eine Kontrolle der GH-Konzentration ist bei der Anwendung von Pegvisomant nicht sinnvoll, da diese aufgrund des Wirkungsmechanismus dieser Substanz erhöht ist, IGF-1 jedoch gesenkt wird. Alternativ zur medikamentösen Therapie kann auch eine radiochirurgische Intervention (Gamma-Knife oder Linearbeschleuniger) zum Ziel führen (Abb.).

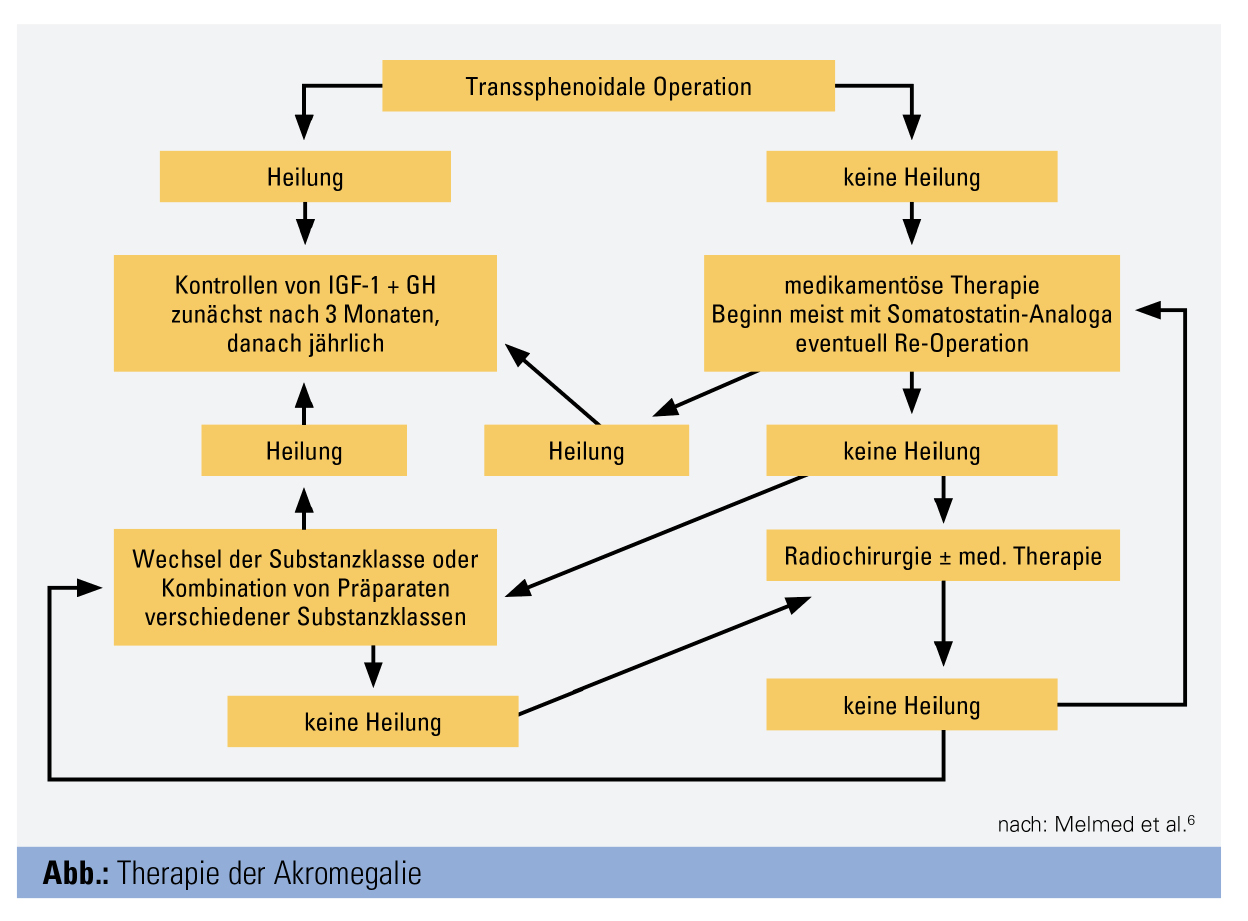
Die Heilung der Akromegalie mit Normalisierung der GH- und/bzw. IGF-1-Konzentrationen stellt das Therapieziel dar, nur dadurch kann die mit Akromegalie assoziierte erhöhte Morbidität und Mortalität beeinflusst werden. Eine konsequente Behandlung der Komorbiditäten, insbesondere jener des Herz-Kreislauf-Systems, ist hierfür ebenfalls von großer Bedeutung. Kontrolluntersuchungen sind zunächst 6 Wochen und 3 Monate nach einer Operation und danach wegen der Rezidiv-Gefahr und zur Überprüfung der übrigen Hypophysen-Funktionen zumindest einmal jährlich über mindestens 10 Jahre erforderlich.
Resümee
Aufgrund der erheblichen Morbidität und zweifach gesteigerten Mortalität ist eine Heilung des GH-Exzesses jedenfalls anzustreben. Die transsphenoidale Entfernung des GH-produzierenden Hypophysen-Adenoms stellt die Therapie der 1. Wahl dar. Wird dadurch keine Heilung erzielt, sind Somatostatin-Analoga der 1. Generation gut wirksame Therapien, die auch die Größe von Tumorresten reduzieren. Weitere therapeutische Alternativen stellen Pegvisomant, Pasireotid und Cabergolin bzw. radiochirurgische Verfahren dar.
1 Jadresic A et al., Q J Med 1982; 51: 189–202
2 Petersenn S et al., Internist 2017; 58: 1171–82
3 McCabe J et al., Neuroendocrinol 2016; 103: 66–74
4 Katznelson L et al., J Clin Endocrinol Metab 2014; 99: 3933–51
5 Casanueva FF et al., Pituitary 2017; 20: 489–98
6 Melmed S et al., Nat Rev Endocrinol 2018; 14: 552–61

AutorIn: Univ.-Prof. Dr. Anton Luger
Klinische Abteilung für Endokrinologie und Stoffwechsel, Universitätsklinik für Innere Medizin III,Medizinische Universität Wien

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases