
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Die Behandlung der thrombotisch-thrombopenischen Purpura (TTP)
24. Mai 2019
Die thrombotisch-thrombopenische Purpura (TTP) ist eine klar definierte Entität einer heterogenen Gruppe von schweren Erkrankungen mit ähnlichen Symptomen, den thrombotischen Mikroangiopathien (TMA). Sie sind durch eine mikroangiopathische hämolytische Anämie mit Erythrozytenfragmentierung, Thrombopenie und unspezifische Organfunktionsstörungen charakterisiert, die vital bedrohlich sein können. Die TTP ist eine seltene Erkrankung, die akut auftritt, dramatisch verläuft und unbehandelt tödlich endet. In den letzten Jahren wurden nicht nur beträchtliche Fortschritte in der Diagnostik von TMA erzielt, sondern auch neue therapeutische Strategien etabliert, die die Prognose deutlich verbessert haben.
Pathophysiologie der TTP
Das gegenwärtige pathophysiologische Konzept versteht die TTP als Zustand eines schweren ADAMTS13-Mangels, der entweder durch genetische Anomalien (angeborene TTP) oder durch Autoantikörper (Autoimmun-TTP) verursacht wird. Das Fehlen von ADAMTS13 führt zur Persistenz von ultragroßen Multimeren des Von-Willebrand-Faktors (UL-VWF MM). In Gegenwart von zusätzlichen Auslösern, die Scherstress verursachen und den VWF entfalten (z. B. Schwangerschaft, Infektionen, bestimmte Medikamente, Operationen usw.), tritt eine verstärkte Aggregation von Thrombozyten an den UL-VWF MM auf. Diese Aggregate beeinflussen den Blutfluss in der Mikrozirkulation, verursachen Organschäden und die klinischen Symptome. Es sind vor allem die besonders vulnerablen Organe Gehirn, Herz und Nieren betroffen.
Die Bestimmung der ADAMTS13-Aktivität ist bei TMA von wesentlicher Bedeutung, da die TTP nur bei sehr geringen Konzentrationen (unterhalb der Nachweisgrenze der meisten Assays) auftritt. Sobald auch nur eine geringe ADAMTS13-Aktivität nachweisbar ist (> 10 % der Norm), muss eine andere TMA-Form vorliegen. Die derzeit verfügbaren Assays, basierend auf ELISAs oder Fluoreszenzmethoden, sind sehr sensitiv, aber nicht allgemein und schnell verfügbar. Vor Kurzem kam jedoch ein Schnelltest auf den Markt (Technoscreen®-ADAMTS13), der innerhalb von 30 Minuten die Diagnose eines schweren ADAMTS13-Mangels ermöglicht.
Angeborener ADAMTS13-Mangel (Upshaw-Schulman-Syndrom): Es sind zahlreiche Mutationen und Polymorphismen im ADAMTS13-Gen bekannt, die zu einer starken Reduktion der ADAMTS13-Aktivität führen. Diese angeborene (familiäre) TTP (OMIM Nr. 274150; http://www.ncbi.nlm.nih.gov/omim) kann sich früh im Kindesalter, aber auch später im Leben (bei Frauen oft während der ersten Schwangerschaft) manifestieren, und Betroffene neigen zu Rückfällen. TTP-Schübe können z. B. durch Infektionen, Schwangerschaft, Operationen, Medikamente etc. ausgelöst werden. Ein internationales Register für Patienten mit Upshaw-Schulman-Syndrom (NCT01257269) sammelt derzeit alle verfügbaren Fälle dieser seltenen Erkrankung.
Erworbener ADAMTS13-Mangel (Autoimmun-TTP): Autoantikörper, die gegen ADAMTS13 gerichtet sind, hemmen entweder dessen Funktion oder erhöhen die Clearance, was zu einem schweren ADAMTS13-Mangel führt. Als Auslöser des Autoimmunprozesses sind verschiedene Faktoren wie Infektionen mit Viren (Epstein-Barr-Virus, Cytomegalovirus, HIV usw.) oder anderen Krankheitserregern, Malignität, bestimmte Medikamente, andere begleitende Autoimmunerkrankungen, Schwangerschaft bekannt. Oft wird aber keine zugrunde liegende Störung gefunden. Der Nachweis von Anti-ADAMTS13-Inhibitoren bestätigt die immunologische Natur.
Klinische Symptome
Die wichtigsten klinischen Symptome von TTP sind, wie bei allen Arten von TMA, die Coombs-negative Hämolyse mit Erythrozytenfragmentierung (erkennbar an Anämie, Erhöhung von LDH, freiem Serum-Hämoglobin, Retikulozyten- und Schistozytenzahl, vermindertem Haptoglobin und Hämoglobinurie), Thrombozytopenie und Zeichen der gestörten Mikrozirkulation.
Die Symptome der Organfunktionsstörungen sind häufig unspezifisch und sehr variabel. Die gestörte Gehirnperfusion kann eine Vielzahl unspezifischer neurologischer Symptome verursachen, die von Kopfschmerzen, verschwommener Sprache, Schwindel oder Agitiertheit bis zu Schlaganfall, Erblindung, epileptischen Anfällen oder Koma reichen. Eine Nierenbeteiligung führt zu erhöhtem Serum-Kreatinin, Oligo- oder Anurie und hämolyseinduzierter Hämoglobinurie. Eine kardiale Beteiligung kann mit einem Anstieg der Herzenzyme, EKG-Veränderungen, Arhythmien, Myokardinfarkt, Kardiomyopathie und akutem Herztod einhergehen. Auch andere Organe (Lunge, Pankreas, Metabolik, Darm) können beteiligt sein. Die Thrombozytopenie geht nur manchmal mit Purpura einher, Blutungen sind selten.
Erstdiagnose
Patienten mit einer akuten TTP-Episode gehören zu den schwierigsten hämatologischen Notfällen. Sofortige geeignete Diagnoseverfahren (Tab. 1) sind erforderlich, um die TTP eindeutig zu identifizieren und sie von anderen TMA-Formen zu unterscheiden. Eine sorgfältige Anamnese wird mögliche Ursachen für die TMA aufdecken. Bevor mit einer Behandlung begonnen wird, sollten Proben von Plasma, Serum und Blutzellen des Patienten gewonnen werden, um für die notwendigen Analysen zu Verfügung zu stehen. Die Beurteilung der Organfunktion sollte mit geeigneten Methoden durchgeführt und die Patienten initial möglichst auf einer Intensivstation betreut werden, da sie sich jederzeit akut verschlechtern können. In jedem Fall sollte bis zur Bestätigung der Diagnose eine TTP vermutet werden und die geeignete Therapie (Tab. 2) ohne unnötige Verzögerung eingeleitet werden, um das beste Ergebnis für die betroffenen Patienten zu erzielen.
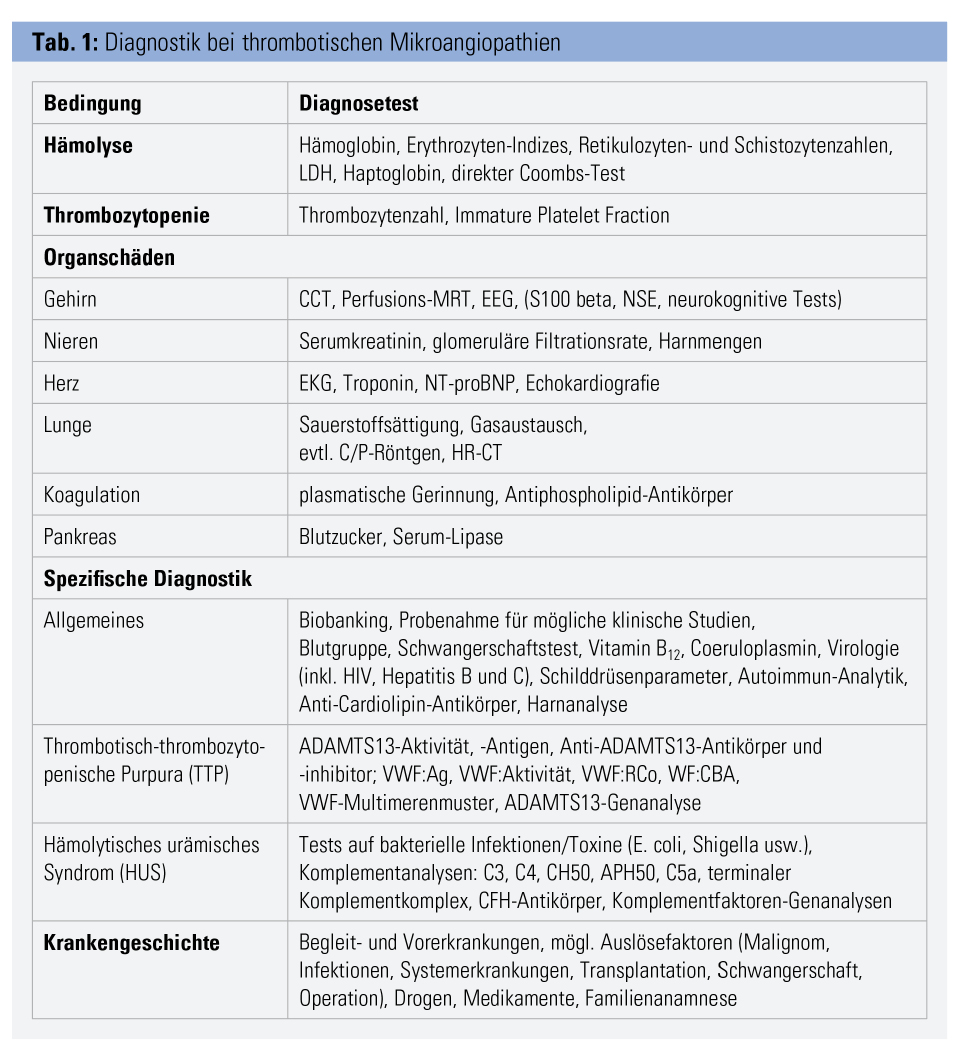
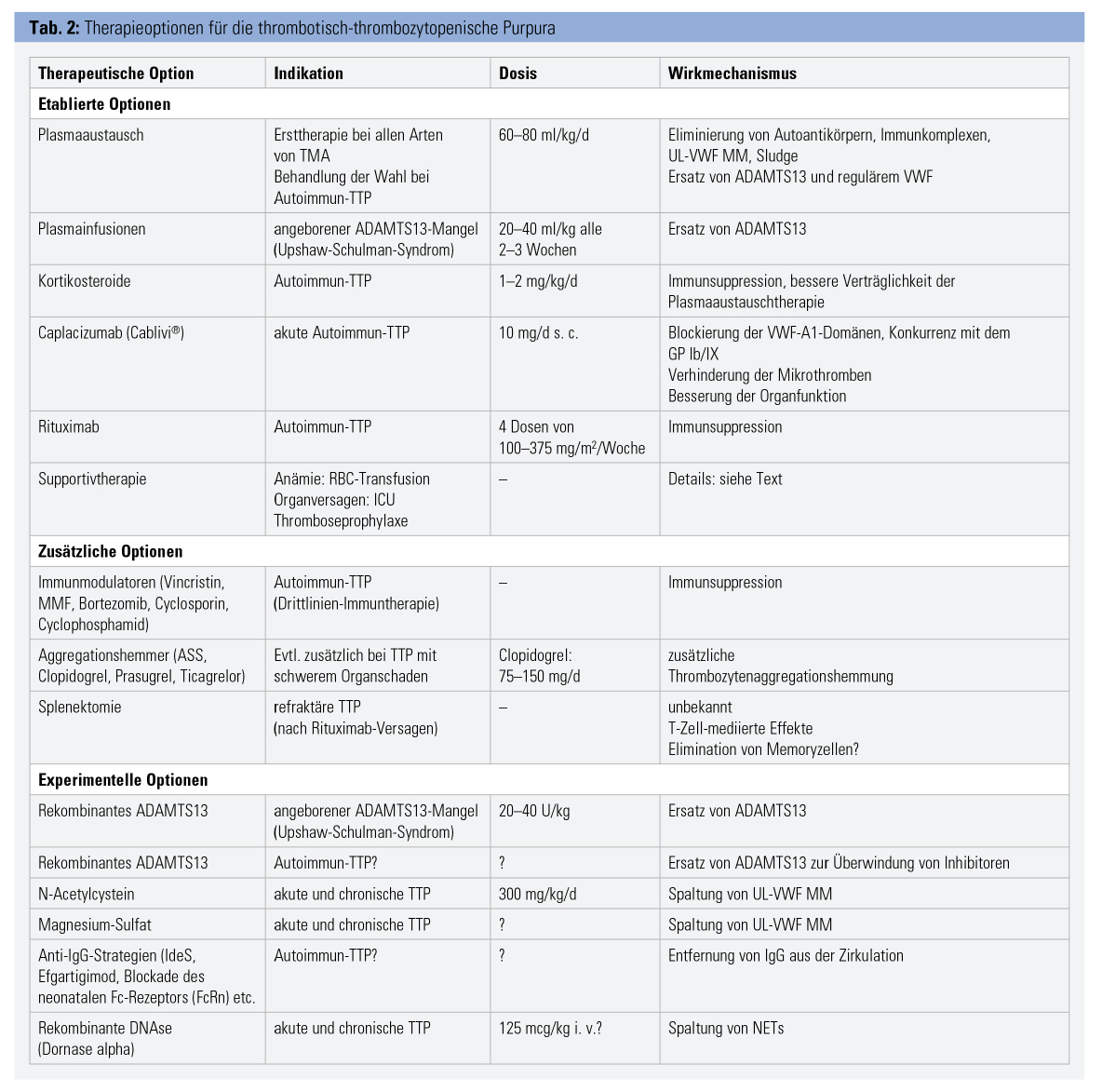
Therapieoptionen
Plasmaaustauschtherapie (PEX)
Durch die PEX wurde das Überleben von TTP-Patienten auf 80–90 % verbessert. Während dieses Verfahrens wird das 1,5-fache Plasmavolumen entfernt und durch Spenderplasma ersetzt. Durch PEX werden Autoantikörper, UL-VWF MM, Immunkomplexe und Sludge entfernt und durch ADAMTS13 sowie VWF mit normaler Multimerenkomposition ersetzt. Die Behandlung muss täglich erfolgen, bis Thrombozytenzahl, LDH, Hämolyse und Organfunktion normalisiert sind. Bei refraktären Fällen und schwerer Organdysfunktion kann die Behandlungsintensität durch Erhöhen des ausgetauschten Plasmavolumens oder durch zweimal tägliche Therapie erhöht werden.
Patienten mit angeborenem ADAMTS13-Mangel sprechen in der Regel schnell an, und die Thrombozytenzahl normalisiert sich innerhalb weniger Tage. Patienten mit sekundären TMA-Typen (transplantassoziierte, einige arzneimittel- oder infektionsinduzierte TMA) sprechen nicht auf PEX an, und die Behandlung sollte nur als symptomatische Maßnahme für Patienten in schlechtem Zustand fortgesetzt werden.
Plasmainfusionen
Die Therapie der angeborenen TTP wird normalerweise durch Plasmainfusionen als Ersatz für das fehlende Enzym durchgeführt. Selbst akute Anfälle mit schweren Symptomen sprechen in der Regel gut auf die Infusion von Plasma an (20–40 ml pro kg Körpergewicht). Bei Patienten mit häufigen Rückfällen oder einer „smoldering TTP“ sind regelmäßige prophylaktische Plasmainfusionen (alle 2–3 Wochen) erforderlich, da diese Patienten häufig eine unterschwellige neurologische Symptomatik aufweisen.
Immunsuppression
Kortikosteroide (1–2 mg/kg KG Prednison tägl.) werden bei Autoimmun-TTP verwendet, um die weitere Antikörperbildung zu unterdrücken, um Akut-Phasen-Reaktionen und den Scherstress zu reduzieren, die endotheliale Funktion zu verbessern und Nebenwirkungen des PEX zu reduzieren. Eine Immunsuppression mit Rituximab ist bei Autoimmun-TTP hoch effektiv (aber außerhalb der Zulassung) und erreicht eine komplette Elimination der Autoantikörper innerhalb weniger Wochen. Die Wirkung hält in der Regel mehr als 2 Jahre an. Eine randomisierte klinische Studie (STAR-Studie; NCT00799773) wurde jedoch aufgrund einer niedrigen Rekrutierungsrate abgebrochen. In Anbetracht des hohen Risikos einer dauerhaften Organschädigung bei rezidivierender oder refraktärer TTP müssen die eventuellen Nebenwirkungen von Rituximab gegen die Chance abgewogen werden, eine langanhaltende Remission zu erzielen. Refraktäre oder häufig rezidivierende Patienten erhalten oft eine Drittlinien-Immunsuppression mit Substanzen wie Vincristin, Mycophenolat-Mofetil (MMF), Cyclophosphamid, Bortezomib oder Bendamustin.
Caplacizumab
Caplacizumab (Cablivi®) ist ein bivalentes Nanobody, das rekombinant aus den funktionellen Fragmenten der schweren Ketten von Lama-Immunglobulinen besteht und die A1-Domänen des VWF (die physiologischen Liganden der Plättchenrezeptoren GP Ib/IX) spezifisch und mit hoher Affinität bindet. Somit konkurriert Caplacizumab mit der Thrombozytenbindung und hemmt daher die Thrombozytenaggregation und -aktivierung, beeinflusst jedoch nicht die Kollagenbindung oder die ADAMTS13-Empfindlichkeit von VWF. Caplacizumab wird subkutan verabreicht und bewirkt eine Unterdrückung der VWF-Funktion für bis zu 48 Stunden nach einer Einzeldosis. Es wurde in zwei randomisierten kontrollierten Studien (TITAN, HERCULES) bei Patienten mit akuter Autoimmun-TTP untersucht und führte zu einer signifikanten Reduktion der Zeit bis zur Normalisierung der Thrombozyten, der Organschäden, der Exazerbationen, des Ressourcenverbrauches und der Mortalität. Caplacizumab ist zur Therapie der akuten Autoimmun-TTP zugelassen und verfügbar. Es wird in einer Dosis von 10 mg nach jedem PEX s. c. verabreicht, danach auch bei schon normalen Thrombozytenzahlen so lange, bis die ADAMTS13-Aktivität wieder nachweisbar ist.
Splenektomie
Die Splenektomie hat auch heute noch eine Berechtigung bei schweren, refraktären oder relapsierenden Verläufen einer Autoimmun-TTP. Der Wirkmechanismus ist unklar, wahrscheinlich spielt die Elimination von Immunzellen (T-Lymphozyten, Memory-Zellen) dabei eine Rolle.
Supportivtherapie
Die Transfusion von Erythrozyten ist notwendig, wenn die Hämolyse zu schwerer Anämie führt, wobei jedoch die optimale Transfusionsschwelle undefiniert ist. Thrombozytentransfusionen sind kontraindiziert, da sie die Mikrothrombenbildung verstärken. Bei TTP-Patienten ist häufig eine intensivmedizinische Behandlung erforderlich, da sich die klinische Situation in kurzer Zeit trotz Therapie akut verschlechtern kann. Eine neurologische Verschlechterung durch zerebrale Ischämie kann eine Sedierung, eine mechanische Beatmung oder eine antiepileptische Therapie erforderlich machen. Eine kardiale Minderperfusion erfordert eine hämodynamische Überwachung, Kreislaufunterstützung oder koronare Interventionen. Nierenversagen erfordert ein sorgfältiges Flüssigkeits- und Elektrolytmanagement und häufig eine Nierenersatztherapie. Während all dieser anspruchsvollen medizinischen Behandlungen müssen PEX und spezifische TTP-Behandlungen fortgesetzt werden, was zu logistischen Herausforderungen führen kann. Eine Thromboseprophylaxe (z. B. mit niedermolekularen Heparinen) ist auch bei niedrigeren Thrombozytenzahlen notwendig.
Neue therapeutische Optionen
Rekombinantes ADAMTS13: Derzeit wird rekombinantes ADAMTS13 (rADAMTS13) zur Substitutionstherapie bei kongenitalem ADAMTS13-Mangel entwickelt. Es wird wahrscheinlich einen großen Fortschritt bei der Behandlung der kongenitalen TTP darstellen, da damit eine Plasmatherapie mit allen Nachteilen (zeitaufwändiges Auftauen, großes Flüssigkeitsvolumen, potenzielles Risiko der Übertragung von Krankheitserregern, Nebenwirkungen, Immunogenität, allergische Reaktionen) unnötig und eine prophylaktische oder therapeutische Heimbehandlung (wie bei Hämophilen) möglich sein wird. Es besteht jedoch ein gewisses Risiko der Entwicklung von Anti-ADAMTS13-Alloantikörpern.
Es sind auch Studien mit rADAMTS13 bei Autoimmun-TTP geplant, wo versucht werden soll, durch hohe rADAMTS13-Dosen die Auswirkung der Autoantikörper hintanzuhalten.
N-Acetylcystein, Magnesium-Sulfat: N-Acetylcystein (NAC) ist eine antioxidative Substanz, die seit vielen Jahren klinisch als Mukolytikum verwendet wird. In Modellen mit Humanplasma, gereinigtem VWF- und ADAMTS13-Knock-out-Mäusen konnte gezeigt werden, dass NAC zu einer Verringerung von UL-VWF MM führt, indem die Disulfidbindungen im VWF gespalten werden. Eine klinische Studie zur Anwendung bei TTP läuft.
Ähnlich soll Magnesiumsulfat wirken, auch damit wird eine aktive klinische Studie in Frankreich durchgeführt.
Anti-IgG-Strategien: Methoden zur Reduktion des Plasma-IgG-Spiegels oder zur Blockade von Autoantikörpern werden bei verschiedenen Autoimmunerkrankungen seit Langem mit wechselndem Erfolg verwendet und auch bei der Autoimmun-TTP eingesetzt. Dazu zählen die Plasmapherese, extrakorporale Immunadsorption oder auch die Infusion von hochdosierten IgG-Konzentraten.
Zurzeit werden aber auch neue Verfahren entwickelt, die zum Teil auch schon bei der TTP verwendet wurden oder ein hohes therapeutisches Potenzial haben könnten. Dazu zählen IdeS, Efgartigimod oder die Blockade des neonatalen Fc-Rezeptors (FcRn) mit spezifischen Antikörpern. Dabei ist jedoch zu beachten, dass die Autoantikörper auch anderen Immunglobulin-Subklassen angehören können (IgM, IgA).
Abschließende Bemerkungen
Bei der Diagnose und Therapie der TTP wurden in den letzten Jahren beträchtliche Fortschritte erzielt. Neue Assays zur Messung der ADAMTS13-Aktivität und von Anti-ADAMTS13-Antikörpern können die Diagnose (Unterscheidung zwischen angeborener und autoimmuner TTP und den verschiedenen anderen TMA-Typen) innerhalb weniger Stunden festlegen und sind hilfreich für die Behandlung. Die darauffolgende PEX-Therapie hat die Überlebensrate der Autoimmun-TTP deutlich verbessert, zusätzlich sind Caplacizumab, Rituximab und Steroide Bestandteile des Therapiekonzepts. Die kongenitale TTP spricht gut auf den Ersatz von ADAMTS13 durch Plasmainfusion an.
Trotzdem darf die Erkrankung nicht unterschätzt werden, sie kann dramatisch verlaufen. Das intensivmedizinische Management ist komplex und erfordert viel Expertise, um die logistischen Hindernisse und die interdisziplinäre Kooperation koordinieren zu können.
Therapeutischer Ausblick
Die nahe Zukunft wird wahrscheinlich Änderungen der Behandlungskonzepte und weitere therapeutische Ansätze mit sich bringen, die eventuell sogar ohne PEX auskommen: Ersetzen von ADAMTS13 durch ein rekombinantes ADAMTS13-Konzentrat, Spaltung von VWF mit NAC, bessere immunsuppressive Strategien oder Elimination von IgG. Die geringe Inzidenz der TTP erschwert die Durchführung randomisierter kontrollierter Studien mit einer ausreichenden Anzahl von Patienten. Auch weltweit laufende Studien leiden unter schlechter Rekrutierung, was die Entwicklung neuer Therapien teuer und zeitaufwendig macht. Dies ermutigt die behandelnden Ärzte, individuelle Off-Label-Behandlungen durchzuführen, wenn das therapeutische Prinzip in das pathophysiologische Verständnis der Erkrankung passt. Der Schweregrad der Erkrankung und das hohe Risiko, bleibende Schäden zu entwickeln, rechtfertigen eindeutig solche Ansätze in einigen Situationen.
Literatur beim Verfasser

AutorIn: Ao. Univ.-Prof. Dr. Paul Knöbl
Klinische Abteilung für Hämatologie und Hämostaseologie, Universitätsklinik für Innere Medizin I, Medizinische Universität Wien,Karl-Landsteiner-Institut für seltene Erkrankungen in der Hämatologie, Wien

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases