


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Die hereditäre Transthyretin-Amyloidose
24. Mai 2019
Die Erstbeschreibung von Amyloidablagerungen geht auf den deutschen Pathologen Rudolf Vyrchow zurück, der sie bereits 1858 in seiner Publikation „Die Cellularpathologie“ erwähnte.1 Amyloide sind falsch gefaltete Proteine, die über ihre Cross-β-Struktur definiert sind.2 Diese spezielle Tertiärstruktur bedingt Änderungen in den biochemischen Eigenschaften der betroffenen Proteine, was zur Bildung von Amyloidfibrillen und schließlich zu Amyloidplaques führt.
Amyloid lagert sich, mit wenigen Ausnahmen, im Extrazellularraum verschiedener Organe ab und kann so zu deren Dysfunktion führen. Dies wird als Amyloidose bezeichnet. Die Internationale Amyloidose-Gesellschaft listet derzeit 36 Proteine, die beim Menschen eine Amyloidose verursachen können.3
Die häufigsten und somit klinisch relevantesten Formen der Amyloidose im Menschen sind die Alzheimer-Demenz, die Leichtketten-Amyloidose, die Serum-Amyloid-A-Amyloidose und die Transthyretin-Amyloidose (ATTR). Da in dieser Arbeit der Fokus auf ATTR liegt, muss bezüglich anderer Amyloidoseformen auf einschlägige Literatur verwiesen werden.4
Pathophysiologie
Transthyretin (TTR) ist ein Transportprotein für Vitamin A und Schilddrüsenhormone, das vorwiegend in der Leber (> 95 %), aber auch im Plexus choroideus sowie im Auge synthetisiert wird. Bei der ATTR lässt sich eine erblich bedingte Form (mTTR) von einer nichterblich bedingten Form (Wild-Type-ATTR) unterscheiden.1 Bezüglich weiterer Informationen, welche die Wild-Type-ATTR betreffen, muss an dieser Stelle wiederum auf einschlägige Literatur verwiesen werden.5
Die mATTR ist eine autosomal dominant vererbte Erkrankung, bei der es aufgrund von Punktmutationen im TTR-Gen zu einer Destabilisierung des TTR-Tetramers und somit zu einer Dissoziation in seine Monomere kommt. Da nur die Monomere Amyloidfibrillen formen können, stellt dieser Prozess der Dissoziation auch einen Schlüsselmechanismus in der Pathophysiologie der ATTR dar.6
Klinische Präsentation
Die klinische Präsentation der Patienten hängt naturgemäß davon ab, welche Organe von den Amyloidablagerungen betroffenen sind. Die vorwiegend betroffenen Organe bei der mATTR sind das Herz sowie das periphere Nervensystem. Man konnte jedoch auch TTR-Ablagerungen in Nieren, Gastrointestinaltrakt, zentralem Nervensystem sowie Augen betroffener Patienten finden. In welchen Organsystemen sich die mATTR manifestiert, hängt wiederum von der zugrundeliegenden Mutation ab, von denen bisher 140 verschiedene beschrieben wurden.7, 8
Kardiologischer Phänotyp: Zu den Mutationen, die mit einem vorwiegend kardiologischen Phänotyp vergesellschaftet sind, zählen V122I, I68L, L111M, T60A.7
Pathophysiologisch kommt es aufgrund der myokardialen Amyloidablagerung und der daraus resultierenden erhöhten Myokardsteifigkeit zur Ausprägung einer Herzinsuffizienz mit vorwiegend diastolischer Dysfunktion. Diese ist durch Belastungsdyspnoe, Beinödeme, Hepatomegalie, Aszites und einen erhöhten Jugularvenendruck gekennzeichnet.6 Ebenso können Patienten aufgrund von Ablagerungen in den Koronargefäßen an Angina Pectoris leiden. Amyloidablagerungen im Reizleitungssystem des Herzens manifestieren sich teils als lebensbedrohliche Rhythmusstörungen.9
Neurologischer Phänotyp: Die neurologische Symptomatik bei ATTR resultiert primär aus einer pathologischen extrazellulären Amyloidablagerung an Fasern des peripheren und autonomen Nervensystems. Darüber hinaus kann es seltener auch zu einer Anreicherung an den Meningen oder in den Gefäßwänden zerebraler Arterien und Arteriolen kommen.10 Der neurologische Phänotyp der mATTR ist durch die unterschiedliche Verteilung der Ablagerungen bei unterschiedlichen Mutationen relativ heterogen, aber von primär sensorischer Polyneuropathie und autonomer Dysregulation geprägt, wobei die ersten Symptome in der Regel die längsten Nerven betreffen.11 Die Erkrankung beginnt zumeist mit einer sensiblen Neuropathie in den Zehen und breitet sich nach proximal aus, wobei rasch auch motorische Funktionen betroffen sein können.12 Charakteristisch ist eine ausgeprägte Dysautonomie, mit Symptomen wie Dyshidrose, sexueller Dysfunktion oder gastrointestinalen Beschwerden.13 Eine weitere häufige Manifestation ist ein oft bilaterales Karpaltunnelsyndrom.14
Bemerkenswert erscheint, dass selbst bei Trägern der gleichen Punktmutation, beispielsweise Val50Met, sowohl das Alter bei Erkrankungsbeginn als auch die Ausprägung und der Verlauf der Symptomatik unterschiedlich sein können.15 Als ursächlich für diese Unterschiede, die sich auch regional ausprägen, werden verschiedene Mechanismen wie Antizipation oder genetische Modifikatoren diskutiert.16, 17
Die mATTR-Polyneuropathie (mATTR-PNP) hat eine generell schlechte Prognose, die allerdings je nach zugrundeliegender Mutation deutlich unterschiedlich ausfallen kann. Letztlich führen bei allen Erkrankten die fulminante autonome Entgleisung sowie eine generalisierte Kachexie zum Tod.14, 18
Diagnose
Der wichtigste Schritt in der Diagnosestellung einer ATTR ist, diese Erkrankung differenzialdiagnostisch in Betracht zu ziehen.
Für eine korrekte und rasche Diagnostik ist eine gute interdisziplinäre Zusammenarbeit zwischen Neurologie, Kardiologie, Nephrologie, Onkologie, Hämatologie, Gastroenterologie, Radiologie, Nuklearmedizin, Pathologie sowie Genetik unabdingbar. Um diese Zusammenarbeit realisieren zu können, wurde zum Beispiel an der Medizinischen Universität Wien ein eigenes Amyloidoseboard ins Leben gerufen.
Diagnosestellung aus kardiologischer Sicht: Seitens der Kardiologie ist die Diagnosefindung sehr von bildgebenden Untersuchungsmodalitäten dominiert. Eine wichtige Rolle nimmt hierbei aufgrund ihrer breiten Verfügbarkeit die Echokardiografie ein. Sogenannte „Red Flags“, die auf eine kardiale Amyloidose hinweisen können, sind eine bi-ventrikuläre Hypertrophie, ein vor allem basal reduzierter Strain („Apical Sparing“) sowie ein hypertrophiertes interatriales Septum. Häufig findet sich auch ein Perikarderguss.
Eine weitere hilfreiche Untersuchung stellt die kardiale Magnetresonanztomografie dar. Insbesondere „Late-Enhancement“-Aufnahmen und T1-Mapping können hier zur Diagnosefindung beitragen.19, 11
Die wichtigste nichtinvasive bildgebende Untersuchungsmethode zur Diagnose einer kardialen ATTR ist jedoch die Knochenszintigrafie mit radioaktiv markierten Tracern wie 99mTc-markiertem 3,3-diphosphono-1,2-propanodicarboxylic acid (Abb.).10
Neben bildgebenden Verfahren stellen Serum- und Urinanalysen die zweite diagnostische Säule dar. Diese können einerseits wichtige Informationen über das zugrunde liegende Amyloid-Protein und andererseits über mögliche Organbeteiligungen geben.6, 10 So kann ein Paraprotein in Serum- oder Harnimmunfixation hinweisend auf eine leichtkettenassoziierte Amyloidose sein. Erhöhte (NT-pro-)BNP- sowie Troponin-Werte können wiederum auf eine kardiale Mitbeteiligung im Rahmen einer Amyloidose deuten.12
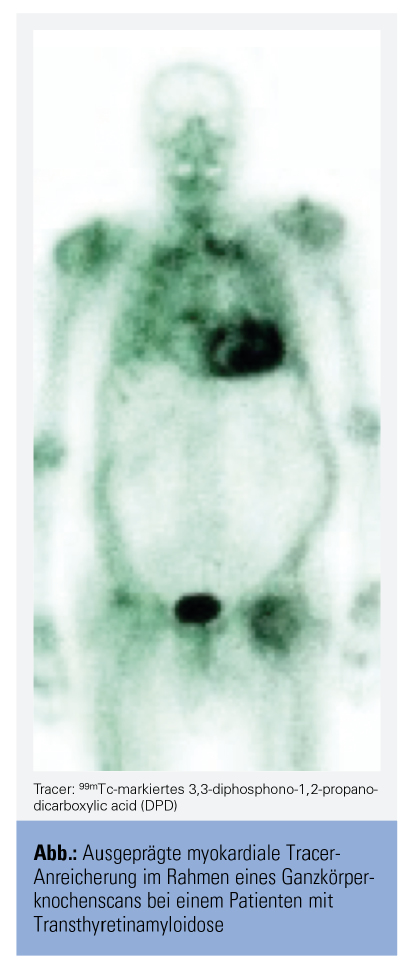
Den kritischsten Punkt in der Amyloidosediagnostik stellt aufgrund der therapeutischen Konsequenzen die Differenzierung der ATTR von anderen Amyloidosen, insbesondere der Leichtketten-Amyloidose dar. Durch einen von Gillmore et al. 2016 in Circulation publizierten Algorithmus ist nun in vielen Fällen bereits ohne Gewebebiopsie eine korrekte Diagnosefindung möglich. Sollte sich in Serum- und Harnanalysen kein Hinweis auf ein Paraprotein finden sowie eine ausgeprägte myokardiale Tracer-Anreicherung im Ganzkörperknochenscan vorhanden sein, ist bei dieser Befundkonstellation keine Biopsie notwendig (positiver Vorhersagewert für das Vorliegen einer ATTR ist 100 %).6 Bei anderen beziehungsweise unklaren Befundkonstellationen oder speziellen Fragestellungen gilt die Biopsie (am besten aus dem betroffenen Organ) nach wie vor als Goldstandard in der Amyloidosediagnostik.6
Diagnosestellung aus neurologischer Sicht: Die frühe Diagnose ist aus neurologischer Sicht gerade bei jungen Patienten aufgrund der klinischen Heterogenität eine Herausforderung, für den Verlauf aber von größter Bedeutung. Wie bei vielen seltenen Erkrankungen ist die Verzögerung vom Beginn der Erkrankung bis zur Diagnosestellung auch bei mATTR relativ groß.11 An die Diagnose einer mATTR sollte gedacht werden, wenn eine bilaterale und symmetrische Polyneuropathie rasch progredient ist, zur Einschränkung der Gehfähigkeit führt und eines der folgenden Kriterien erfüllt ist: positive Familienanamnese, autonome Dysregulation, kardiale oder renale Beteiligung, unerklärter Gewichtsverlust oder bilaterales Karpaltunnelsyndrom.20
An diagnostischen Methoden können einerseits etablierte Techniken wie Nervenleitgeschwindigkeit oder Elektromyografie zur Bestätigung der Polyneuropathie und zum Ausschluss einer anderen Diagnose zum Einsatz kommen. Spezifischer für die mATTR sind experimentelle diagnostische Methoden wie zum Beispiel MR-Neurografie oder Messung der elektrochemischen Leitfähigkeit der Haut.21, 22 Eine Biopsie kann aus subkutanem Fettgewebe, Speicheldrüsen oder betroffenen Nerven erfolgen. Eine negative Biopsie schließt jedoch eine mATTR nicht aus.23–25 So ist letztlich die genetische Testung (s. u.) bei klinischem Verdacht der entscheidende diagnostische Schritt aus neurologischer Sicht.26
Genetische Analyse: Sobald die Diagnose einer ATTR gestellt wurde, muss, um zwischen mATTR und Wild-Type-ATTR unterscheiden zu können, eine Analyse des TTR-Gens angeboten werden. Diese darf jedoch nur nach ausführlicher Aufklärung über die möglichen Konsequenzen einer positiven genetischen Untersuchung, die therapeutischen Konsequenzen und schriftlichem Einverständnis des Betroffenen durchgeführt werden.
Therapie
Lange Zeit war bei der mATTR eine Organtransplantation betroffener Organe bzw. der Leber als TTR-Produktionsort die einzige therapeutische Option. Durch intensive Bemühungen gibt es nun seit 2018 bereits drei für die mATTR zugelassene medikamentöse Therapieoptionen.
Dieses Kapitel soll einen Überblick über supportive sowie spezifische Therapiemöglichkeiten der mATTR-PNP sowie der kardialen mATTR geben.
Therapie aus neurologischer Sicht
Derzeit gibt es drei amyloidmodifizierende therapeutische Ansätze bei der mATTR-PNP. Die TTR-Stabilisierung, die Supprimierung der TTR-Synthese und die Elimination von bereits abgelagertem TTR-Amyloid. Darüber hinaus hat bei jungen Patienten mit gewissen Mutationen eine Lebertransplantation nach wie vor ihren Stellenwert.27
TTR-Stabilisatoren: In diese Substanzklasse fällt das bereits seit 2013 zugelassene Tafamidis, das zu einer Verlangsamung des Fortschreitens der PNP führt.28 Dieses Medikament bindet an das TTR-Tetramer, stabilisiert dieses und verhindert so dessen Dissoziation in Monomere, womit Tafamidis in einen Schlüsselmechanismus der ATTR-Pathophysiologie eingreift.
Supprimierung der TTR-Synthese: Ein weiterer therapeutischer Ansatz ist es, die TTR-Produktion in der Leber zu supprimieren und damit weitere Ablagerungen zu verhindern bzw. deutlich zu reduzieren. Hier gibt es derzeit zwei Substanzklassen: „small interfering RNA“ ([siRNA], z. B. Patisiran) und Antisense-Oligonukleotide ([AO], z. B. Inotersen). Sowohl Patisiran als auch Inotersen sind in Europa bereits zur Behandlung der mATTR-PNP zugelassen. Unter Therapie mit Patisiran kam es sogar zu einer Verbesserung der PNP, bei Patienten in der Placebogruppe dagegen zu einer Verschlechterung.29 Bei Inotersen zeigte sich in der Zulassungsstudie eine geringere Progredienz der PNP im Vergleich zu Placebo.30
Elimination von abgelagertem Amyloid: Der dritte Ansatz, den Krankheitsprozess bei ATTR medikamentös zu beeinflussen, ist die Elimination von bereits abgelagerten Amyloidplaques. Serum-Amyloid-P-(SAP-)Antikörper scheinen hierbei besonders vielversprechend. Mit einem ersten SAP-Antikörper wird SAP aus der systemischen Zirkulation eliminiert. Im Anschluss wird ein zweiter SAP-Antikörper appliziert der an Amyloidplaques gebundenes SAP für Makrophagen und Komplementsystem markiert.31 Bisher gibt es jedoch aus dieser Substanzklasse noch keine zugelassenen Medikamente.
Supportive Therapien: Aus neurologischer Sicht sollten insbesondere die durch die autonome Dysfunktion auftretenden Beschwerden sowie allenfalls neuropathische Schmerzen aggressiv behandelt werden. Dafür eignen sich in der Regel gängige Präparate, so zum Beispiel Pregabalin bei Schmerzen oder Metoclopramid für gastrointestinale Beschwerden.26
Therapie aus kardiologischer Sicht
Die Therapiemöglichkeiten für Patienten mit kardialer mATTR hängen sehr davon ab, ob auch eine neurologische Beteiligung vorliegt, denn für Patienten mit einer rein kardiologischen mATTR gibt es nach wie vor keine zugelassene Therapie.
Supportive Therapien: Den Grundstein der supportiven Therapie von Patienten mit kardialer Beteiligung im Rahmen einer mATTR bilden Diuretika.32 Allerdings ist auch hier Vorsicht geboten, da betroffene Patienten aufgrund der erhöhten Ventrikelsteifigkeit a priori höhere Füllungsdrücke benötigen, um eine adäquate ventrikuläre Füllung und somit Auswurf zu erzielen.4, 5
Für typische Herzinsuffizienzmedikamente wie Beta-Blocker, Angiotensin-Converting-Enzyme-(ACE-)Hemmer oder Angiotensin-Rezeptor-Blocker gibt es bei der mATTR keine Evidenz.32 In einer kleinen retrospektiven Studie fand man sogar Hinweise darauf, dass deren Einsatz negative Auswirkungen auf die Prognose haben könnte.33 Ein weiterer Faktor bei der Anwendung von Herzinsuffizienzmedikamenten ist die schlechte Verträglichkeit, die der Hypotonieneigung und Notwendigkeit einer Bedarfstachykardie geschuldet ist. Bei fehlender Indikation sollten diese Medikamente daher nach Möglichkeit abgesetzt werden. Sollte tatsächlich eine Bradykardisierung, z. B. im Rahmen eines tachykarden Vorhofflimmerns, notwendig werden, scheinen hier Beta-Blocker und Amiodaron am geeignetsten. Für Digitalispräparate und Kalziumantagonisten gibt es aus In-vitro-Studien Hinweise, dass diese Medikamente an Amyloidfibrillen binden und somit zu lokal toxischen Wirkspiegeln führen können.
Spezifische Therapien: Bis dato gibt es für rein kardiologische Formen der mATTR keine zugelassenen medikamentösen Therapien. Dies wird sich aller Voraussicht nach mit 2020 ändern, wo aufgrund positiver Daten einer Phase-III-Studie mit Tafamidis die erste Therapie für rein kardiale mATTR-Patienten zugelassen werden sollte.34 Ansonsten soll dieser Abschnitt einen Überblick über die Datenlage der unterschiedlichen Substanzklassen bei der mATTR aus kardiologischer Sicht verschaffen.
TTR-Stabilisatoren: Die erwähnte Phase-III-Studie mit Tafamidis wurde von Maurer et al. im New England Journal of Medicine publiziert (ATTR-ACT-Studie).34 Hierbei handelte es sich um eine placebokontrollierte, internationale, multizentrische Studie mit 440 Teilnehmern, die entweder eine kardiale mATTR oder eine kardiale Wild-Type-ATTR hatten. Patienten in der Tafamidis-Gruppe hatten gegenüber den mit Placebo behandelten Patienten ein signifikant besseres Überleben.34 An der Amyloidoseambulanz der Universitätsklinik für Innere Medizin II, Abteilung für Kardiologie der Medizinischen Universität Wien (Leitung: Assoc. Prof. Priv.-Doz. Dr. Diana Bonderman) steht diese Therapie dort betreuten Patienten bereits seit 2 Jahren im Rahmen eines Named-Patient-Programms zur Verfügung.
Supprimierung der TTR-Synthese: Im Rahmen der Zulassungsstudien von Patisiran und Inotersen wurden auch Effekte auf kardiale Parameter untersucht.30, 35, 36 Die Datenlage für Patisiran ist jedoch deutlich besser als für Inotersen.37 Ob und wann diese Präparate bei kardialen Formen der mATTR angewandt werden können, ist derzeit Gegenstand weiterer Studien.
Elimination von abgelagertem Amyloid: Für diesen Therapieansatz gibt es leider bisher keinerlei kardiologische Daten. Da er sehr vielversprechend scheint, werden Stud
ien, die den Einfluss der Elimination von Amyloidablagerungen auf das Myokard untersuchen, mit Spannung erwartet.
Resümee: Die mATTR wird in den nächsten Jahren aufgrund größerer Awareness sowie besserer diagnostischer Methoden mit zunehmender Häufigkeit diagnostiziert werden. Durch neue Therapieoptionen wandelte sich die mATTR von einer unheilbaren zu einer behandelbaren Erkrankung.
1 Virchow R et al., Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre: zwanzig Vorlesungen, gehalten während der Monate Februar, März und April 1858 im pathologischen Institute zu Berlin 1858
2 Meinhardt J et al., Der Pathologe 2009; 30:175–81
3 Sipe JD et al., Amyloid 2016; 23:209–13
4 Gertz MA et al., Nat Rev Cardiol 2015; 12:91–102
5 Gertz MA et al., J Am Coll Cardiol 2015; 66:2451–66
6 Coelho T et al., Neurol Ther 2016; 5:1–25
7 Rapezzi C et al., Eur Heart J 2013; 34:520–8
8 Rowczenio DM et al., Hum Mutat 2014; 35:E2403–12
9 Rochlani Yogita M et al., Circulation 2015; 132:A20097-A
10 Sekijima Y. Hereditary Transthyretin Amyloidosis. In: Adam MP, Ardinger HH, Pagon RA et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2019
11 Plante-Bordeneuve V et al., Lancet Neurol 2011; 10:1086–97
12 Hund E., Appl Clin Genet 2012; 5:37–41
13 Ando Y et al., Amyloid 1998; 5:288–300
14 Carr AS et al., J Neurol Neurosurg Psychiatry 2016; 87:620–7
15 Koike H et al., Neurology 2004; 63:129–38
16 Hellman U et al., Amyloid 2008; 15:181–6
17 Santos D et al., Ann Clin Transl Neurol 2016; 4:98–105
18 Koike H et al., J Neurol Neurosurg Psychiatry 2012; 83:152–8
19 Duca F et al., JACC Cardiovasc Imaging 2018; 11(12):1924–6
20 Conceição I et al., J Peripher Nerv Syst 2016; 21(1):5–9
21 Kollmer J et al., Neurology 2017; 89:475–84
22 Castro J et al., Clin Neurophysiol 2016; 127:2222–7
23 van Gameren II et al., Arthritis Rheum 2006; 54:2015–21
24 Dardiotis E et al., Amyloid 2009; 16:32–7
25 Koike H et al., Amyloid 2011; 18:53–62
26 Ando Y et al., Orphanet J Rare Dis 2013; 8:31
27 Holmgren G et al., Lancet 1993; 341:1113–6
28 Coelho T et al., Neurology 2012; 79:785–92
29 Adams D et al., N Engl J Med 2018; 379:11–21
30 Benson MD et al., N Engl J Med 2018; 379:22–31
31 Richards DB et al., N Engl J Med 2015; 373:1106–14
32 González-López E et al., Rev Esp Cardiol (Engl Ed):991–1004
33 Aus dem Siepen F et al., Amyloid 2017; 24:132–3
34 Maurer MS et al., N Engl J Med 2018; 379:1007–16
35 Adams D et al., BMC Neurol 2017; 17(1):181
36 Benson MD et al., Amyloid 2017; 24(Sup 1):134–5
37 Dasgupta NR et al., JACC 2018; 71(11 Supplement): DOI: 10.1016/S0735-1097(18)31201-4

AutorIn: Dr. Franz Duca, PhD
Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

AutorIn: Dr. Raphael Wurm
Universitätsklinik für Neurologie, Medizinische Universität Wien

AutorIn: Dr. René Rettl
Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

AutorIn: Dr. Christina Binder
Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

AutorIn: Assoz. Prof. Priv.-Doz. Dr. Diana Bonderman
Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases
