


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Mukopolysaccharidosen
24. Mai 2019
Erhebliche kumulative Prävalenz: 1 von 8.000
Mukopolysaccharidosen (MPS) können unbehandelt zu schwerster körperlicher und geistiger Behinderung führen. Das klinische Bild und der Verlauf dieser seltenen angeborenen Stoffwechselerkrankungen variiert zwischen verschiedenen Formen und sogar individuell erheblich, ist jedoch meist von skelettalen, viszeralen und in etwa 70 % der Patienten von ZNS-Manifestationen gekennzeichnet. Die Heterogenität des klinischen Bildesund die Seltenheit der einzelnen MPS-Formen führen dazu, dass Patienten häufig zu spät diagnostiziert werden. Kausale Therapien und eine adäquate Betreuung werden dadurch unnötig verzögert. Die kumulative Prävalenz aller Formen ist mit ca. 1 von 8.000 erheblich, sodass auch Ärzte außerhalb spezialisierter Zentren mit den wichtigsten Fakten zu MPS vertraut sein sollten.
Ursache und Einteilung
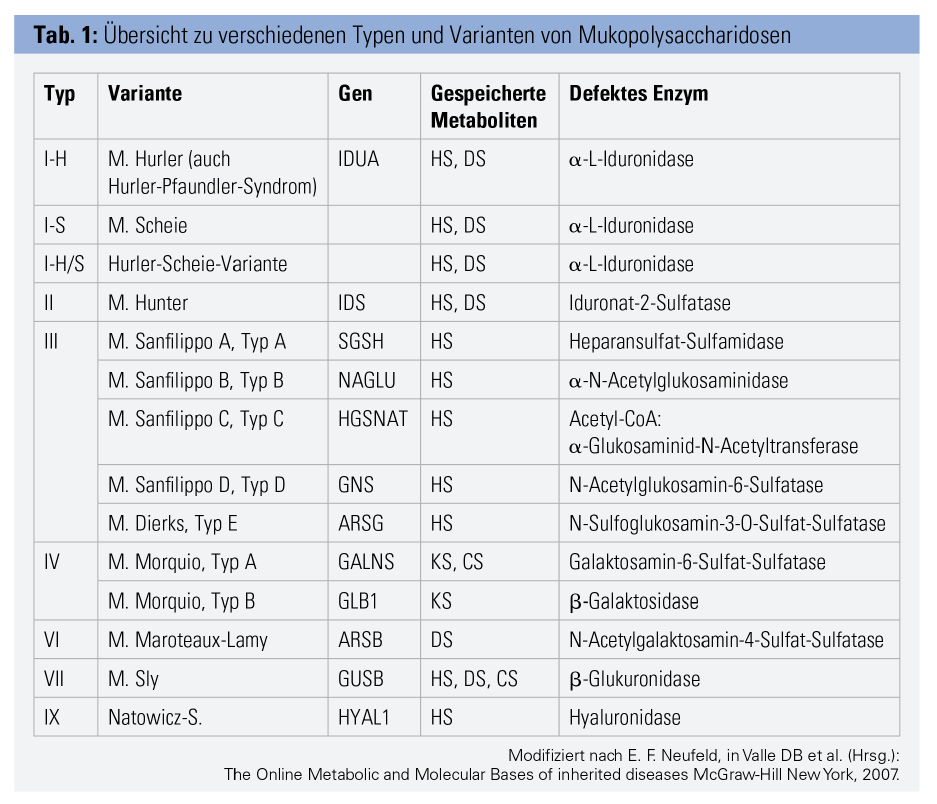
Genetisch bedingte Defekte lysosomaler Enzyme führen zu einem verminderten Abbau von Glykosaminoglykanen (kurz GAGs, früher „Mucopolysaccharide“) aus der Bindegewebsmatrix. Je nachdem, welches der 12 GAG-abbauenden Enzyme betroffen ist, kommt es zu einer der 9 MPS-Formen bzw. mit Sub-Formen zu 14 verschiedenen Erkrankungen (Tab. 1). Es handelt sich um monogenetisch x-chromosomal (Typ 2) oder autosomal-rezessiv (alle anderen Formen) vererbte Krankheiten.
Das klinische Bild und Differenzialdiagnosen
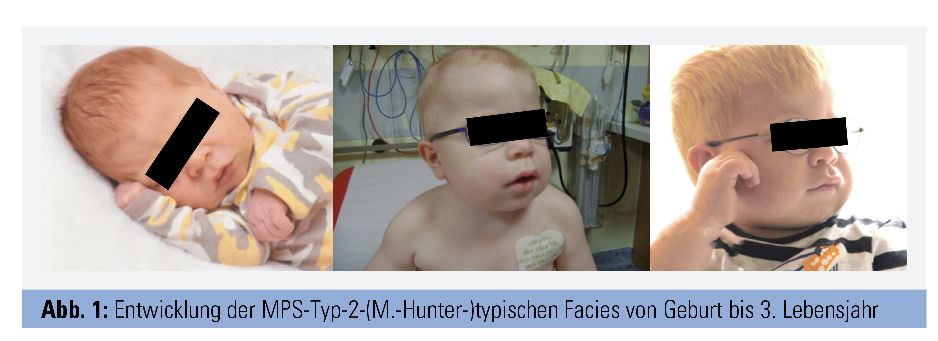
Alle Formen zeigen einen chronisch progredienten Verlauf. Bei Geburt wirken die Kinder meist gesund, die Akkumulation von GAGs führt jedoch im Verlauf zu zunehmender Vergrößerung („Speicherorgane“) und/oder Störung von Struktur und Funktion der Organe (Abb. 1 und 2). Betroffen sind vor allem Skelett, ZNS und eine große Anzahl anderer Organsysteme. Abbildung 3 fasst die häufigsten Organmanifestationen zusammen. Welche Organe betroffen sind, unterscheidet sich je nach MPS-Form, aber auch individuell. Die Typen I, II, VI und VII sind meist von einer Vielzahl von Symptomen in Skelett und mehreren anderen Organen gekennzeichnet, wobei die schweren Verläufe von Typ I, II und VII zu kognitiven Störungen und Verhaltensauffälligkeiten führen. Bei den Typ-IV-Formen stehen schwere Skelettveränderungen bei normaler Intelligenz im Vordergrund, während Typ-III-Patienten vor allem von kognitiven Defiziten, Verhaltensstörungen und z. T. Epilepsie betroffen sind. Zudem reicht das Spektrum von sehr schwer verlaufenden frühkindlichen Formen bis zu spät manifestierenden attenuierten Formen. MPS betreffen daher nicht nur das Kindes-, sondern auch das Erwachsenenalter. Sehr ähnlich können sich andere lysosomale Speicherkrankheiten wie Mukolipidosen und Oligosaccharidosen (z. B. die behandelbare Alpha-Mannosidose) präsentieren.
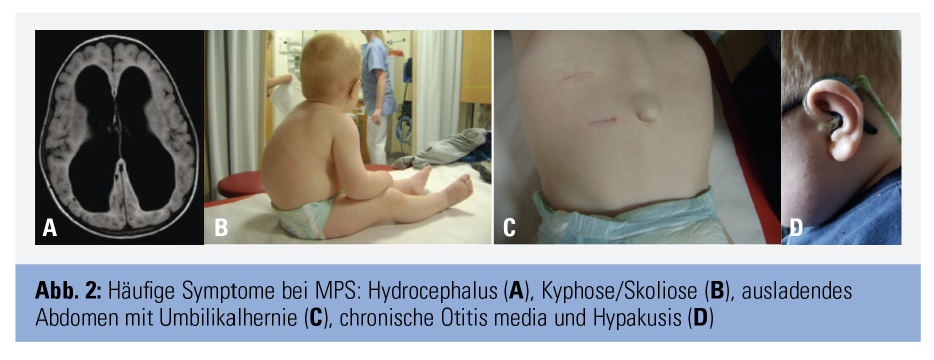
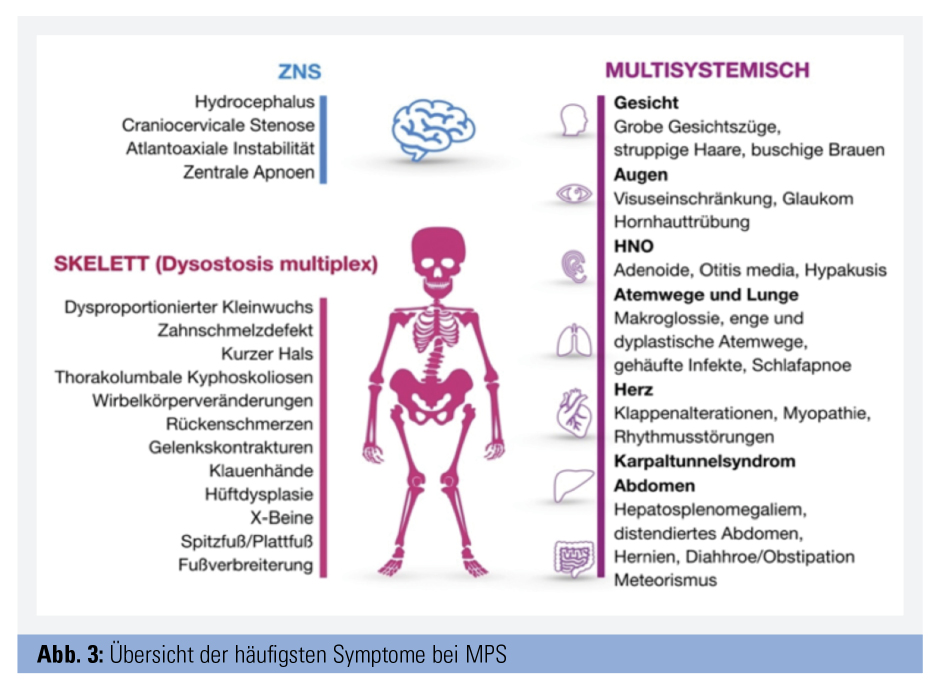
Diagnose: typische Symptommuster
Die Diagnose MPS gelingt durch Nachweis der erniedrigten Enzymaktivität und der pathogenetischen Mutationen mittels weniger Tropfen Blut auf einer Filterkarte. Die eigentliche Herausforderung besteht darin, an MPS als Verdachtsdiagnose zu denken. Nur wenige der vielen möglichen Symptome sind einzeln betrachtet charakteristisch, wie z. B. die klassische Facies mit struppigen Haaren, buschigen Augenbrauen und vergröberten Gesichtszügen, das Karpaltunnelsyndrom im Kindesalter und die sogenannten Klauenhände, welche durch Verdickung und Kontrakturen der Fingergelenke entstehen. Fallen diese Zeichen auf, ist die MPS jedoch häufig schon deutlich fortgeschritten. Es gilt daher, charakteristische Kombinationen von Symptomen zu erkennen, um klassische Formen rechtzeitig bzw. attenuierte Formen überhaupt der gezielten Diagnostik zuzuführen. So ist beispielsweise die zufällige Koinzidenz einer kongenitalen Kyphose (ca. 1 von 1.000) und eines Hydrocephalus (ca. 1 von 1.000) mit etwa 1 von 1.000.000 anzunehmen und damit etwa zehnfach seltener als die beim in Tabelle 2 beschriebenen Patienten dafür ursächliche MPS vom Typ 2.
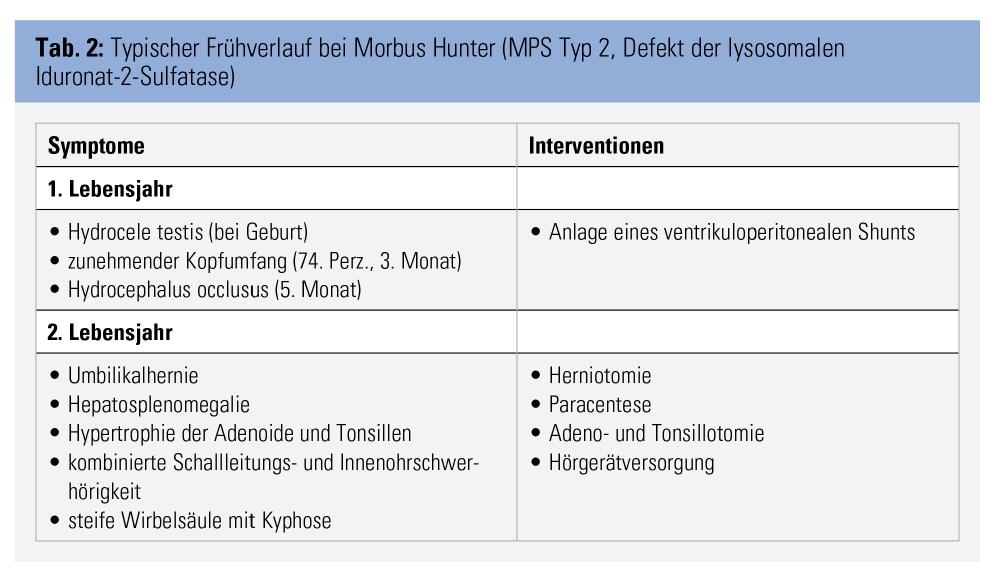
Andere MPS-typische Muster, die eine diagnostische Abklärung triggern sollten, sind z. B. multiple Hernien und Skelett- bzw. Gelenkanomalien, Hernien und Hypertrophie der Adenoide und Tonsillen, Hepato-Splenomegalie und eines oder mehrere der oben genannten Symptome. Es ist dabei nicht notwendig, dass man suspekte Symptomkombinationen gezielt der MPS oder einer anderen der 6.000 bis 8.000 seltenen Erkrankungen selbst zuordnen kann, da hierfür spezialisierte Zentren für seltene Erkrankungen mit interdisziplinären Experten-Boards kontaktiert werden können. Entscheidend ist jedoch, zu reagieren, wenn Patienten multiple Probleme präsentieren, die nicht schlüssig erklärt werden können.
Therapie: Möglichkeiten und Herausforderungen
Für die MPS-Typen I, II, IV A, VI und VII stehen Enzymtherapien zur Verfügung, die als wöchentliche intravenöse Infusionen verabreicht werden. Obschon es sich um kausale Therapien handelt, kann dadurch keine Heilung, sondern lediglich eine Verbesserung der Symptome bzw. eine verlangsamte Progression erzielt werden. Wird die Diagnose zu spät gestellt, liegen häufig schon erhebliche irreversible Schäden vor. Auch Patienten mit einer MPS-Form, für die noch keine Therapie zugelassen ist, profitieren von der korrekten Diagnose, da sie so einer geeigneten Betreuung zugeführt werden können. Neben den Expertenzentren spielen Selbsthilfegruppen eine wichtige Rolle dabei, Patienten und ihren Familien eine optimale Versorgung zu ermöglichen. Die Gesellschaft für Mukopolysaccharidosen in Österreich wurde 1984 als Verein gegründet (www.mps-austria.at) und bietet den betroffenen Familien nicht nur die Möglichkeit zum Austausch, sondern darüber hinaus umfassende Unterstützung in vielen Aspekten.
Fazit
Etwa 1 von 8.000 Personen sind von MPS betroffen. Gelingt es, suspekte Symptommuster zu erkennen, kann die Diagnose durch Bestimmung der Enzymaktivität und des Genotyps gestellt und die Betreuung durch ein gut abgestimmtes, interdisziplinäres Team übernommen werden. Für die MPS vom Typ I, II, IV, VI und VII sind kausale Therapien verfügbar. Weitere Arzneimittel werden aktuell entwickelt. Neben spezialisierten Zentren spielen Selbsthilfegruppen eine wichtige Rolle für die optimale Betreuung der Patienten.

Priv.-Doz. Dr. Florian B. Lagler
Universitätsklink für Kinder- und Jugendheilkunde,Institut für angeborene Stoffwechselerkrankungen, Paracelsus Medizinische Privatuniversität Salzburg

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases
