


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Thalassämie – eine der häufigsten Erbkrankheiten
24. Mai 2019
Alle Hämoglobintypen des Menschen sind aus zwei Paaren von Globinketten aufgebaut, bestehen also aus vier Untereinheiten. Jede Globinkette enthält als prosthetische Gruppe ein Häm mit einem zentralen Eisen (FeII).
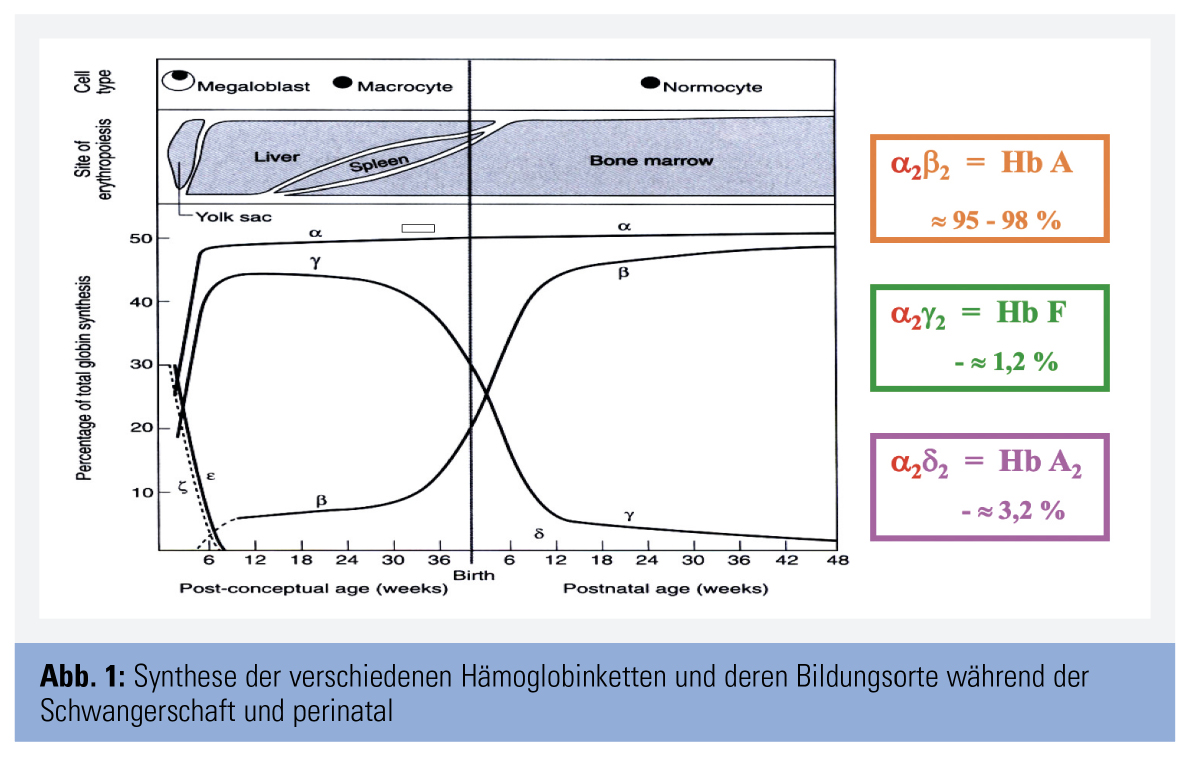
Zunächst werden embryonale Hämoglobine unter Beteiligung von - und -Ketten gebildet; die Expression dieser Ketten sistiert gegen Ende der Embryonalperiode. In der anschließenden Fetalzeit werden andere Hämoglobine synthetisiert: Hämoglobin A (Hb A), Hämoglobin F (Hb F) und Hämoglobin A2 (Hb A2). Hb A besteht aus 2 α- und 2 β-Ketten (α2β2), Hb F aus 2 α- und 2 γ-Ketten (α2γ2) und Hb A2 aus 2 α- und 2 δ-Ketten (α2δ2). Die Bildungsrate der unterschiedlichen Globinketten und damit der verschiedenen Hämoglobine ändert sich im Rahmen der Entwicklung. Zunächst überwiegt Hb F; mit zunehmender Reifung nimmt die Hb-F-Synthese ab und die von Hb A steigt an. Etwa ein Jahr post partum ist das Hämoglobinmuster weitgehend stabilisiert; es besteht aus den folgenden Anteilen: Hb A etwa 95–98 %, Hb F bis zu 1,2 % und Hb A2 etwa 2,0–3,2 % (Abb. 1). Diese Verteilung bleibt lebenslang erhalten.
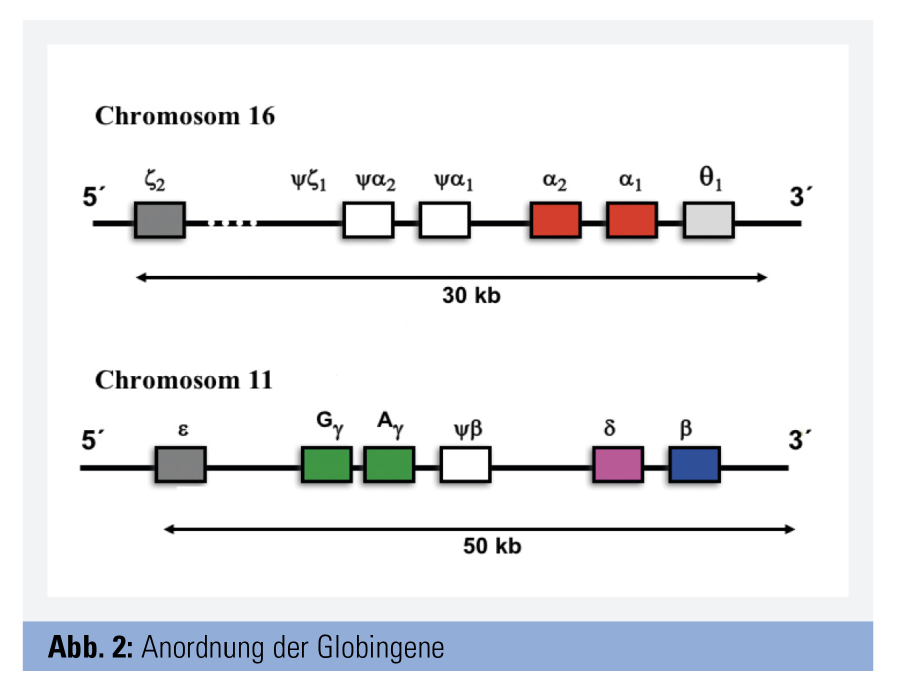
Die Genloci für die Synthese der Globinketten finden sich auf den Chromosomen 11 und 16.
Am Chromosom 11 befindet sich der sogenannte β-Globin-Cluster. Dieser enthält u. a. je einen Genlocus für die β-Kette und die δ-Kette sowie zwei für die γ-Ketten (γA und γG). Die beiden γ-Ketten unterscheiden sich in nur einer Aminosäure und sind physiologisch gleichwertig. Am Chromosom 16 finden sich zwei Loci für die α-Ketten (α2- und α1-Locus). Jeder Gesunde verfügt somit über insgesamt 4 α-Loci. Die beiden α-Gene unterscheiden sich geringfügig in ihren Introns, nicht aber in den Exons, sodass alle α-Loci eine idente Globinkette codieren. Im Normalfall ist die durch das α2-Gen exprimierte Menge an α-Ketten etwa 2–3-mal größer als die vom α1-Gen (Abb. 2).
Epidemiologie
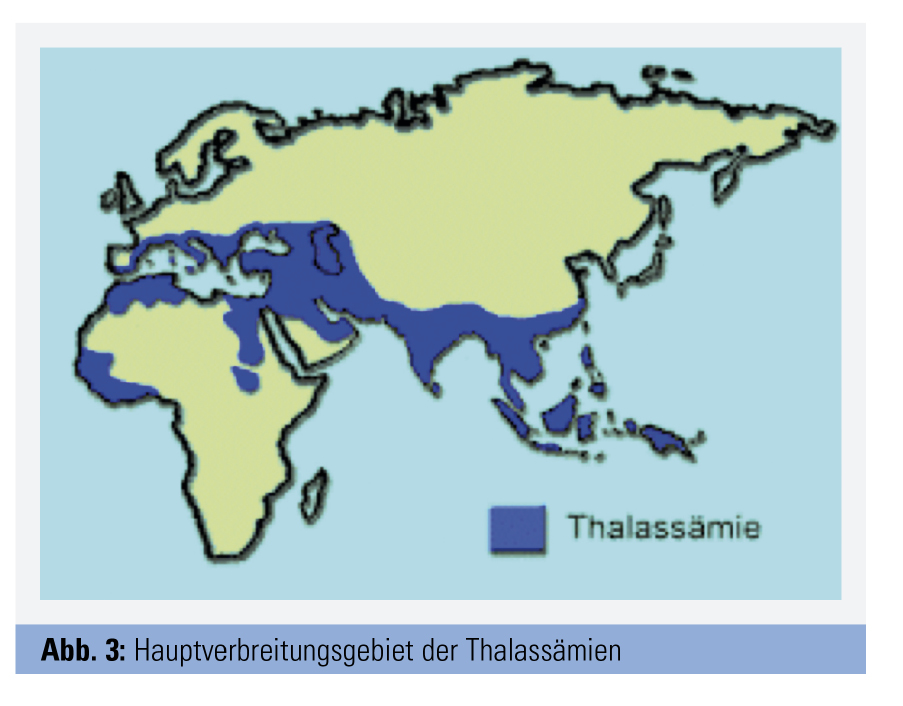
Es wird geschätzt, dass zwischen 4,5 % und 7 % der Weltbevölkerung Träger eines Hämoglobinopathie-Gens sind. Hämoglobinopathien zählen somit global zu den häufigsten genetischen Erkrankungen (Abb. 3). In Mitteleuropa waren Hämoglobinopathien früher selten, doch ist die Zahl von Genträgern in den letzten Jahrzehnten im Zusammenhang mit den modernen Migrationsbewegungen stark im Zunehmen begriffen. Für Österreich gibt es keine genauen Zahlen, mit Blick auf Deutschland (etwa 400.000 betroffene Personen) kann man aber eine Zahl von rund 40.000 Genträgern vermuten.
Bei den Hämoglobinopathien kann man zwei große Gruppen unterscheiden, Hämoglobinvarianten und Thalassämien.
Hämoglobinvarianten enthalten Globinketten mit abnormaler Sequenz. Gegenwärtig sind über 1.300 Varianten beschrieben. Thalassämien sind hingegen Erkrankungen, bei denen der Aufbau der Globinketten normal ist, aber aufgrund von Mutationen zu wenige (+ Thalassämien) oder keine (0 Thalassämien) Globinketten eines bestimmten Typs für die normale Hämoglobinbildung zur Verfügung stehen.
Die verbreitetsten Thalassämie-Formen betreffen Mutationen der α-Kette und β-Kette (α- und β-Thalassämien), doch sind auch für die γ- und δ-Globinketten entsprechende Defekte beschrieben. Insgesamt sind gegenwärtig über 500 Thalassämie-Mutationen bekannt; in diese Zahl inkludiert sind auch 50 Hämoglobinvarianten, bei denen die Mutation sowohl eine Änderung der Globinkettenstruktur als auch ein Defizit bei der Menge der codierten abnormalen Globinkette zur Folge hat.
Alpha-Thalassämien
Verbreitung: α-Thalassämien sind vermutlich die weltweit verbreitetsten Einzelgen-Erkrankungen. Zurzeit sind etwa 128 molekulare Defekte beschrieben, die zu einer α-Thalassämie führen. Die Frequenz an Genträgern unterscheidet sich je nach Weltgegend erheblich. Die höchsten Genfrequenzen (60–80 %) finden sich in Südostasien, Indien und Saudi-Arabien. Im Mittelmeerraum liegt die Frequenz bei 5–10 %; in Nord- und Mitteleuropa war die Frequenz ursprünglich niedrig, nimmt aber im Rahmen der modernen Migrationsbewegungen laufend zu.
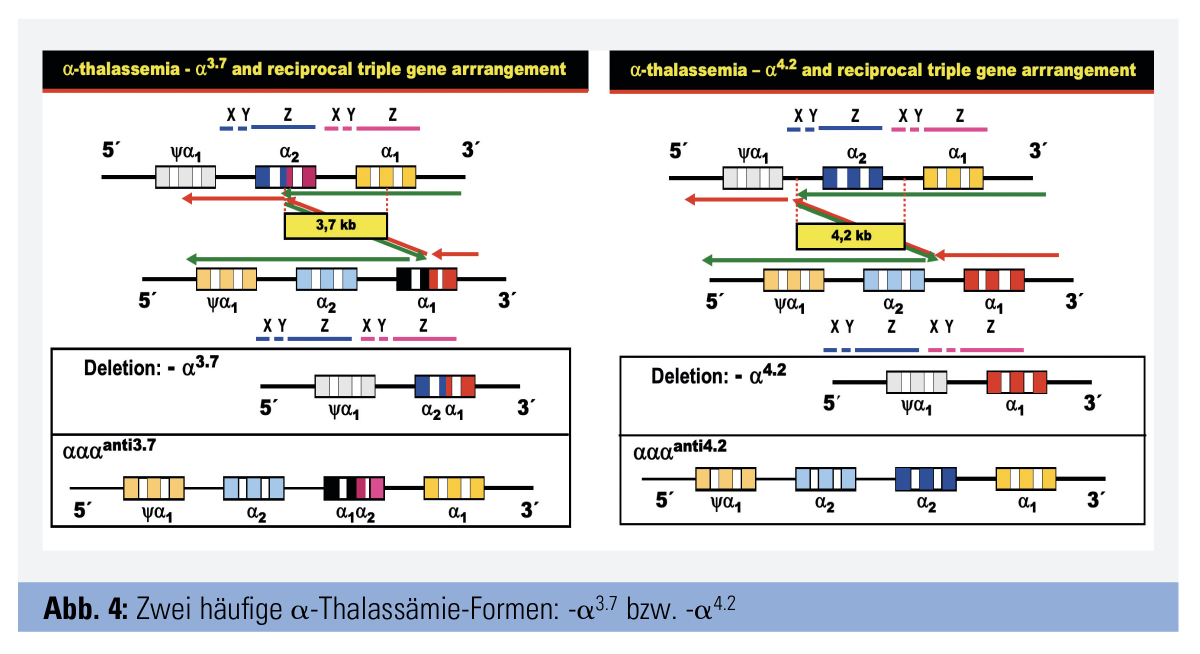
Formen: Die bei weitem häufigsten α-Thalassämie-Formen gehen auf Deletionen zurück, wesentlich seltener sind Punkt-Mutationen. Sehr verbreitet sind zwei kleine Deletionen, die auf Grund eines pathologischen Crossing over zum Verlust eines 3,7 kb bzw. 4,2 kb großen DNA-Segmentes (-α3.7 bzw. -α4.2) führen. Folge ist, dass ein betroffenes Chromosom 16 jeweils nur ein und nicht – wie normal – zwei funktionelle α-Globingene aufweist. Dadurch wird die α-Kettenproduktion verringert, aber nicht eliminiert; der Zustand wird als α+–Thalassämie bezeichnet. Das Crossing over bedingt auch, dass es auf dem homologen Chromosom 16 zu einer Insertion und dadurch zu einem zusätzlichen α-Gen kommt, ein Zustand, der als αααanti 3.7 bzw. αααanti4.2 bezeichnet wird (Abb. 4). Des Weiteren gibt es eine verbreitete Gruppe von α-Thalassämien, die auf verschiedene ausgedehnte Deletionen zurückgehen, die zum Verlust beider α-Gene auf demselben Chromosom 16 führen. Beispiele sind die südostasiatische Deletion (–SEA), die Mediterrane Deletion (–MED), die philippinische Deletion (–Fil) u. a.
Es gibt auch seltene genetische Erkrankungen, die neben dem Bild einer α-Thalassämie durch verschiedene andere Störungen gekennzeichnet sind. Dazu zählen das ATRX-Syndrom und das ATR-16-Syndrom. Das ATRX-Syndrom geht auf Mutationen des ATRX-Gens am X- Chromosom (Xq21.1) zurück und ist u. a. durch ausgeprägte mentale Retardierung, eine charakteristische Erscheinungsform des Gesichtes und Skelettdeformationen charakterisiert. Das ATR-16-Syndrom wird auf unterschiedliche Anomalien am Chromosom 16 (16p13.3) zurückgeführt. Damit verbunden können verschiedene Störungen sein, darunter mentale Retardierung, Gesichtsmissbildungen und Fußdeformitäten (Klumpfuß).
Vereinzelt sind auch erworbene α-Thalassämie-Formen im Zusammenhang mit myeloischen Erkrankungen (α-Thalassemia-Myelodysplasia) beschrieben, die sich in Form einer HbH-Erkrankung (siehe unten) manifestieren. Im Allgemeinen sind sie mit erworbenen Punkt-Mutationen im ATRX-Gen assoziiert. Betroffen sind vorwiegend ältere Männer.
Klinik: Der Verlust von einem oder von zwei funktionellen α-Genen ist klinisch weitgehend unauffällig und zeigt sich im Blutbild als milde mikrozytäre hypochrome Anämie. Bei Verlust zweier α-Gene ist formal zu unterscheiden, ob je ein α-Gen auf beiden Chromosomen 16 fehlt (homozygoter α+-Thalassämie-Trait) oder ob auf einem Chromosom 16 beide α-Gene deletiert sind (heterozygoter α0-Thalassämie-Trait). Der Verlust von drei funktionellen α-Genen führt zur sogenannten HbH-Erkrankung. Bei diesem Zustand bilden die im erheblichen Überschuss vorliegenden β- oder γ-Ketten pathologische afunktionelle Tetramere, die als Hb H (β4) bzw. als Hb Bart’s (γ4) bezeichnet werden. In den meisten Fällen ist eine HbH-Erkrankung durch eine kombinierte Heterozygotie (Compound Heterozygosity) für eine kleine und eine große α-Thalassämie-Deletion bedingt. Die Erkrankung kann auch resultieren, wenn sich auf einem Chromosom 16 eine Punkt-Mutation auf einem der α-Gene befindet und am anderen Chromosom 16 beide α-Gene infolge einer großen Deletion fehlen. Ein relativ häufiges Beispiel ist die –SEA-Mutation kombiniert mit Hb Constant Spring (–SEA/αCSα). Beim Hb Constant Spring wird wegen einer Punkt-Mutation des Stop-Codons im α2-Locus sowohl die gebildete mRNA instabil als auch die in geringer Menge exprimierte abnormale α-Globinkette. Schließlich ist beschrieben, dass auch Homozygotie für eine Punkt-Mutation der Poly-A-Site am α2-Gen (AATAAAAATAAG) eine HbH-Erkrankung verursacht.
Bei der HbH-Erkrankung ist die α-Globinkettenproduktion meist auf unter 30 % des Normalen vermindert. Charakteristisch ist eine Anämie variablen Schweregrads, oft verbunden mit einer Splenomegalie und gelegentlich mit Hypersplenismus. Hinzu können Infektionen und andere Komplikationen sowie Folgeerscheinungen der Hämolyse kommen wie Ikterus, Gallensteine und (seltener) Eisenüberladung.
Beim Hydrops fetalis Bart’s sind alle vier α-Loci deletiert oder inaktiviert. Folge ist, dass in utero die normale Umstellung der Hämoglobinproduktion auf Hb F, Hb A und Hb A2 nicht möglich ist, da alle diese Hämoglobine α-Ketten enthalten. Meist überlebt der Fetus mit schweren Bildungsstörungen bis ins 2. oder 3. Trimenon, weil die Bildung eines embryonalen Hb (Hb Portland) persistiert, und verstirbt noch in utero oder kurz nach der Geburt.
Beta-Thalassämien
Verbreitung: Etwa 1,5 % der Weltbevölkerung sind Träger eines β-Thalassämie-Gens. Global gesehen ist das Verbreitungsgebiet der β-Thalassämien mit dem der α-Thalassämien weitgehend identisch. Die höchsten Genfrequenzen in Europa finden sich im Mittelmeerraum und zwar besonders in den östlichen Anrainerstaaten. So sind in Portugal, Spanien und Frankreich etwa 1–2 % der Bevölkerung betroffen, in Italien etwa 4–5 %, in Griechenland 8 % und auf Zypern 15 %. In Nordafrika finden sich Frequenzen zwischen etwa 3 % und 9 %. In der Türkei beträgt die Durchschnittsfrequenz etwa 2,1 %, allerdings mit deutlichen regionalen Unterschieden.
Formen: Im Gegensatz zu den α-Thalassämien werden β-Thalassämien meist durch Punkt-Mutationen verursacht sowie durch kleine Deletionen und kleine Insertionen im β-Globingen und in unmittelbar „upstream“ oder „downstream“ davon liegenden DNA-Abschnitten.
Je nachdem, ob die β-Globinkettensynthese vermindert oder komplett eliminiert ist, spricht man von β+- oder β0-Thalassämien. Insgesamt sind an die 400 β-Thalassämie-Mutationen beschrieben. Bezüglich der Frequenz der häufigsten Mutationen gibt es deutliche regionale Unterschiede.
Klinik: Personen mit heterozygoter β-Thalassämie (β-Thalassämie-Trait) sind asymptomatisch. Das Blutbild zeigt typischerweise eine mikrozytäre hypochrome Anämie. Die Erythrozytenzahl ist oft deutlich erhöht, kann aber auch im Normalbereich liegen. Das Ferritin ist zwar im Mittel höher als bei Gesunden, übersteigt aber nur selten die Normalwertgrenze.
Auch Neugeborene mit homozygoter oder doppelt heterozygoter β-Thalassämie sind wegen ihres zunächst noch hohen Hb-F-Anteils asymptomatisch. Mit dem physiologischen Absinken von Hb F kommt es dann aber im Allgemeinen zur Ausbildung einer schweren Anämie. Nach dem Erstbeschreiber wird der Zustand auch als Cooley-Anämie bezeichnet. Im Blutausstrich finden sich die typischerweise zahlreichen Schießscheibenzellen (Target Cells). Das Knochenmark ist hyperplastisch. Der Anteil von Hb F am Gesamthämoglobin ist erhöht, bei homozygoter β0-Thalassämie findet sich kein Hb A und das Gesamthämoglobin besteht – abgesehen von einem geringen Anteil an Hb A2 – nur aus Hb F. Pathophysiologisch spielen nicht nur die ineffektive Erythropoese per se eine Rolle, sondern besonders die im relativen Überschuss gebildeten normalen α-Ketten. Denn diese bilden Aggregate, die mit dem Aufbau der Erythrozytenmembran interferieren und zum Untergang erythropoetischer Vorstufen im Knochenmark führen; in der Peripherie führen die Aggregate zu einer verminderten Verformbarkeit der Erythrozyten und beschleunigen so ihre Phagozytose durch das retikuloendotheliale System. In der Folge kommen weitere Pathomechanismen zum Tragen, wie oxidative Schädigung, Hyperkoagulabilität und nicht zuletzt Organschäden. Allerdings gibt es auch mildere Verlaufsformen, die als Thalassemia intermedia bezeichnet werden. Ursache kann das Zusammentreffen der Anlagen für zwei (milde) β+-Thalassämien sein, des weiteren Mutationen, die zu einer Hb-F-Erhöhung führen oder auch eine zusätzlich bestehende α-Thalassämie, da diese zur Folge hat, dass der pathogene Überschuss an α-Ketten weniger ausgeprägt ist.
Neben der „klassischen“ homozygoten β-Thalassämie gibt es eine Reihe von Zuständen, die phänotypisch dem Bild einer β-Thalassämie entsprechen. Dazu zählt z. B. die Kombination einer β-Thalassämie-Anlage mit Heterozygotie für Hb E. Ursache ist, dass die Hb-E-Mutation nicht nur zur Bildung eines abnormalen Hämoglobins führt, sondern dass auch die Bildungsrate der abnormalen Globinkette deutlich vermindert ist (Alternative Splicing). Eine andere Ursache sind Lepore-Hämoglobine. Es handelt sich dabei um Hämoglobine, bei denen anstelle von β-Ketten Hybridketten aus Anteilen der δ- und der β-Globinkette gebildet werden. Die Kombination mit einer β-Thalassämie-Anlage oder Homozygotie für Lepore-Hämoglobine können dem Bild einer homozygoten β-Thalassämie entsprechen.
Eine weitere Gruppe von Zuständen, die dem Formenkreis der Thalassämien zugerechnet werden, sind teils sehr große Deletionen am Chromosom 11, die mehrere Globingene in unterschiedlichem Ausmaß betreffen. In Abhängigkeit der betroffenen DNA-Region kann es zu einem thalassämischen Bild mit Hb-F-Erhöhung (δβ- und γδβ-Thalassämien) oder zu einer asymptomatischen Hb-F-Erhöhung (hereditäre Persistenz von Hb F, HPFH) kommen. Die Übergänge zwischen asymptomatischer Hb-F-Erhöhung und dem klinischen Bild einer Thalassämie sind fließend und hängen von der Art der Deletion ab.
Therapie der Thalassämie
Alpha-Thalassämie: Personen mit α-Thalassämie-Trait benötigen keine Behandlung. Auch bei der HbH-Erkrankung ist meist keine spezifische Therapie erforderlich, allerdings sollte auf ausreichenden Impfschutz geachtet werden. Dies gilt besonders für Kinder nach Splenektomie und auch, weil durch Infektionen oder Medikamente Hämolysen induziert werden können. Die meisten Patienten mit HbH-Erkrankung benötigen keine oder nur vereinzelt Bluttransfusionen. Bei erhöhten Retikulozytenwerten infolge eines gesteigerten Erythrozytenumsatzes kann Folsäure- und Eisensubstitution indiziert sein. Gelegentlich sind orthopädische Eingriffe erforderlich, um Skelettdeformationen, die auf erythropoetisch-hyperplastische Veränderungen zurückgehen, zu korrigieren. Bei schweren Verlaufsformen mit häufiger Gabe von Erythrozyteninfusionen bzw. erhöhtem Ferritin ist eine Eisen-Chelat-Therapie (siehe unten) zu überlegen. Eine Splenektomie wird meist nur bei Hypersplenismus durchgeführt. Eine Heilung ist zurzeit nur durch eine Stammzelltransplantation möglich, eine solche ist aber nur selten indiziert.
Beta-Thalassämie: Personen mit β-Thalassämie-Trait sind nicht behandlungsbedürftig. Ausnahme sind gelegentlich Schwangere, die eine ausgeprägte Eisenmangelanämie entwickeln können. Homozygote und doppelt heterozygote β-Thalassämien können dagegen unbehandelt eine schwere Symptomatik aufweisen. Haupttodesursache ohne begleitende Eisen-Chelat-Therapie sind kardiale Komplikationen aufgrund einer transfusionsbedingten Hämochromatose des Herzens. Andere häufige Komplikationen sind endokrine Störungen, Thromboembolien, Osteoporose und auch Knochenveränderungen bedingt durch die gesteigerte Blutbildung.
Eine allogene Stammzelltransplantation ist bei Vorhandensein eines geeigneten Spenders heute die Therapie der Wahl. Die Transplantation soll möglichst in der Kindheit erfolgen, bevor manifeste Organschäden auftreten. Falls nicht transplantiert werden kann und bei milderen Verlaufsformen (Thalassemia intermedia), besteht das Standardvorgehen in der regelmäßigen Gabe von Erythrozytenkonzentraten und einer begleitenden Eisen-Chelat-Therapie. Zur Überwachung der Organ-Eisenüberladung soll die Magnetresonanztomografie der Ferritinmessung überlegen sein.
Zurzeit stehen für die Eisen-Chelat-Therapie drei Medikamente zur Verfügung: ein älteres Präparat, Deferoxamin (Desferal®), das parenteral verabreicht werden muss, sowie zwei neuere oral wirksame Präparate, Deferipron (Ferriprox®) und Deferasirox (Exjade®).
Mit Ende März 2019 hat die European Medicines Agency die Zulassung einer Gentherapie zur Behandlung der β-Thalassämie (Zynteglo®) empfohlen; diese Empfehlung ist vorläufig auf Personen über 12 Jahre mit transfusionspflichtiger Nicht-β0/β0-Thalassämie beschränkt, für die kein geeigneter Spender zur Verfügung steht. Dabei werden hämatopoetischen Stammzellen des Patienten mit Hilfe eines lentiviralen Vektors funktionelle Kopien eines modifizierten β-Globingens (βA-T87Q) ex vivo eingesetzt und die Zellen dann retransfundiert.
Ein konservativer Therapieansatz ist die Gabe von Hydroxycarbamid (Hydroxyharnstoff), ein oral wirksames Zytostatikum, das auch Hb-F-Bildung auslöst. Mit diesem Präparat konnte bei einem Teil der Patienten mit Thalassemia intermedia eine deutliche Verringerung der Transfusionshäufigkeit erzielt werden. Andere die γ-Kettenproduktion anregende Substanzen sind nur in kleineren Studien untersucht. Weiters hat ein gentechnischer Ansatz die Erhöhung der Hb-F-Produktion zum Ziel, nämlich die Ausschaltung von BCL11A, eines Proteins, das durch Bindung an die γ-Ketten-Promotoren die γ-Kettensynthese hemmt und in der normalen Entwicklung die Umstellung der Hb-F- zur Hb-A-Bildung auslöst.
Einen anderen Therapieansatz verfolgt das in klinischer Prüfung befindliche Präparat Luspatercept (ACE-536); diese Substanz fördert die normale Erythropoese, indem sie GDF11 bindet, einen Liganden, der die Differenzierung erythropoetischer Vorläuferzellen verhindert und so die Symptomatik der Erkrankung verschlechtert.
Grundlagen der Diagnostik
Bei der Untersuchung auf Hämoglobinopathien wird im Allgemeinen eine Stufendiagnostik durchgeführt. Zunächst erfolgt meist eine Untersuchung des Blutbildes, eine quantitative Analyse des Hämoglobinmusters und in unterschiedlichem Ausmaß einige Spezialuntersuchungen, z. B. Stabilitätstests und eine Hb-F-Färbung. Je nach Ergebnis folgt dann eine weitere Differenzierung/Bestätigung auf Protein- und/oder DNA-Ebene. Zur sicheren Diagnose einer α-Thalassämie ist in jedem Fall eine DNA-Untersuchung erforderlich, da die Standard-Screening-Untersuchungen unauffällig sind. Hingegen findet sich bei einer β-Thalassämie-Anlage in den meisten Fällen ein erhöhter Hb-A2-Wert, sodass zur Diagnosestellung oft die Untersuchung auf Proteinebene genügt.
Homozygote β-Thalassämien und doppelt heterozygote Hämoglobinopathien sind meist auf DNA-Ebene abzuklären.
Genetische Beratung/Prävention/Pränataldiagnostik
Screening-Programme für Hämoglobinopathien gibt es auf allen Kontinenten. Diese sind nicht einheitlich. So gibt es Programme für präkonzeptionelle Untersuchungen, Pränataldiagnostik, neonatales Screening, Studentenberatung und voreheliche Beratung. In Europa sind seit mehreren Jahrzehnten vorwiegend regionale Programme etabliert – und zwar in den meisten Mittelmeerländern, einschließlich Frankreich. Aber auch Länder mit ursprünglich niedriger Frequenz an Hämoglobinopathien wie die USA, Kanada, England, die Niederlande, Belgien u. a. sind dazu übergegangen, derartige Programme einzuführen.
In Bezug auf β-Thalassämien kommen prinzipiell alle Paare für eine genetische Beratung bzw. pränatale Diagnostik in Frage, bei denen ein Partner zumindest ein β-Thalassämie-Gen trägt und der andere homo- oder heterozygot für eine das β-Gen betreffende Thalassämie-Form oder eine β-Globinkettenvariante ist.
Bei α-Thalassämien ist ein solches Vorgehen besonders bei Paaren indiziert, bei denen beide heterozygot für eine große Deletion sind oder ein Partner eine große und der andere eine kleine (α-Thalassämie-Trait) Deletion aufweist.
Literatur beim Verfasser

AutorIn: Univ.-Prof. DDr. Pierre Hopmeier
Klinisches Institut für Labormedizin,Universitätsklinikum St. Pölten, Karl Landsteiner Privatuniversität für Gesundheitswissenschaften

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases
