


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Interstitielle Lungenerkrankungen (ILD) können als pulmonale Manifestation im Rahmen von Systemerkrankungen, als Reaktion auf unterschiedliche Noxen oder idiopathisch auftreten.
In manchen Fällen kann es zur Ausbildung einer Lungenfibrose kommen.
Systematik
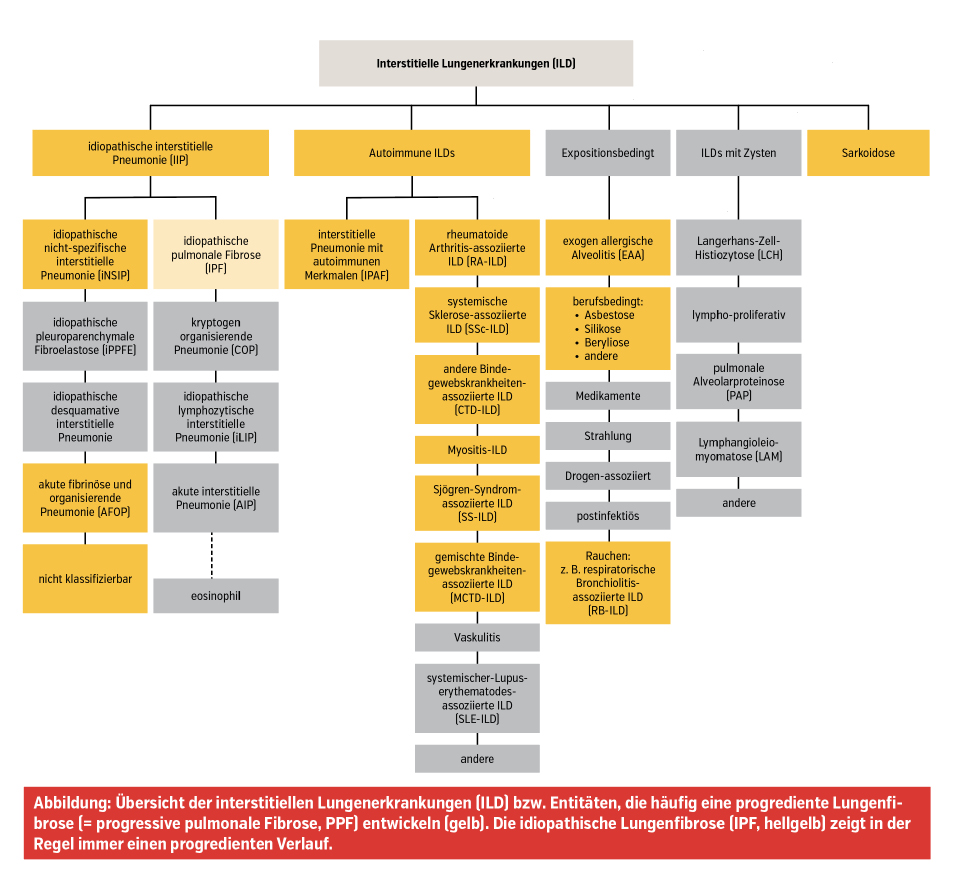
Die Gruppe der ILD kann in unterschiedliche Arten unterteilt werden. Eine aktuell geläufige Systematik ist in der Abbildung dargestellt. Während die idiopathische Lungenfibrose (IPF) praktisch immer zunehmend fibrosierend verläuft und dadurch mit einer sehr schlechten Prognose einhergeht, ist die Wahrscheinlichkeit einer progressiven Lungenfibrose bei den übrigen ILDs unterschiedlich. Kommt es bei ihnen zum Auftreten zunehmender fibrotischer Veränderungen mit begleitendem Abfall der Lungenfunktion, wird dieser Phänotyp unabhängig von der zugrunde liegenden Diagnose auch als progressive pulmonale Fibrose (PPF) bezeichnet. Die Identifizierung einer ILD als PPF ist besonders relevant, da das bei der IPF schon seit längerem eingesetzte antifibrotische Medikament Nintedanib seit kurzem auch bei PPF verordnet werden kann.
Der Weg zur Diagnose
Aufgrund einer unspezifischen Symptomatik werden Patient:innen mit ILD oft erst relativ spät nach Auftreten der ersten Symptome diagnostiziert. Typischerweise stehen trockener Husten sowie zunehmende Atemnot im Vordergrund. Bei der klinischen Untersuchung können Trommelschlägelfinger und Uhrglasnägel auffallen. Die Auskultation der Lunge kann bei Vorliegen von Knisterrasseln einen weiteren Hinweis auf eine Lungenfibrose geben. Bei der Lungenfunktionsüberprüfung liegen typischerweise eine restriktive Ventilationsstörung sowie eine Einschränkung der Diffusionskapazität vor. Bei der Blutgasanalyse (BGA) ist bei fortgeschrittener Erkrankung eine Oxygenierungsstörung zuerst bei Belastung, später auch in Ruhe erkennbar. In frühen Krankheitsstadien können sowohl Lungenfunktion als auch BGA unauffällig sein.
Das Lungenröntgen kann Hinweise auf das Vorliegen einer interstitiellen Strukturvermehrung geben, die weitere Abklärung erfordert jedoch immer eine hochauflösende Computertomografie (HR-CT) der Lunge. Auf Basis der ausführlichen Anamnese, serologischer Untersuchungen (wie z. B. der Suche nach möglichen Autoimmunerkrankungen/rheumatischen Erkrankungen), der klinischen Inspektion und des HR-CT-Befundes sollten in einer interdisziplinären Besprechung eine Diagnose und Behandlung definiert werden. Diese Fallbesprechungen in ILD-Zentren werden klassischerweise von Vertreter:innen der Pneumologie, Rheumatologie, Radiologie und Pathologie abgehalten. Bei unklaren Ergebnissen kann es auch erforderlich sein, eine Lungenbiopsie durchzuführen, die entweder chirurgisch oder neuerdings auch endoskopisch mittels einer transbronchialen Kryobiopsie erfolgen kann.
Therapieoptionen
Die Therapie der ILD richtet sich nach den auslösenden Faktoren und den jeweiligen pathologischen Prozessen. Aufgrund der Komplexität der verschiedenen Erkrankungsformen ist eine Therapieauswahl in einem spezialisierten Zentrum ratsam. Verlaufskontrollen sollten individuell vereinbart werden, dabei kann eine Verknüpfung von Spitals- und niedergelassenem Bereich durchaus sinnvoll sein.
Bei bekanntem Auslöser steht die Beendigung oder Vermeidung der auslösenden Faktoren im Vordergrund, zum Beispiel bei raucherassoziierten Erkrankungen oder medikamentenassoziierter ILD. Die Allergenkarenz spielt bei der exogen-allergischen Alveolitis eine entscheidende Rolle, daher ist die akribische Suche nach einer möglichen Ursache einer ILD essenziell.
Bei der medikamentösen Therapie spielen die immunmodulierende und antifibrotische Therapie eine wesentliche Rolle. Eine immunmodulierende Behandlung sollte zum Beispiel bei Sarkoidose mit Organeinschränkung, bei kryptogener organisierender Pneumonie oder idiopathischer nichtspezifischer interstitieller Pneumonie und anderen ILD erwogen werden. Glukokortikoide stellen in der Regel die Erstlinientherapie dar, der Behandlungszeitraum ist jedoch je nach Erkrankung unterschiedlich. Der Einsatz weiterer Immunsuppressiva zur Kortison-Reduktion ist ebenfalls möglich. Auch Biologika finden zunehmend Anwendung, z. B. bei Vaskulitiden oder therapieresistenten Verlaufsformen. Die IPF sollte jedenfalls frühzeitig mit den antifibrotischen Medikamenten Nintedanib oder Pirfenidon behandelt werden. Aufgrund von Studiendaten hat sich der Anwendungsbereich der Antifibrotika zuletzt deutlich erweitert. Sie werden mittlerweile neben der IPF auch bei PPF sowie der sklerodermieassoziierten ILD angewendet.
Fibrotische Veränderungen sind leider kaum reversibel, sodass die Verhinderung einer fortschreitenden funktionellen Einschränkung das primäre Therapieziel darstellt. Dazu leisten auch die pulmonale Rehabilitation, Langzeitsauerstofftherapie und Schulung der Patient:innen einen wesentlichen Beitrag. Bei bestimmten ILD und bereits deutlicher Einschränkung sollte auch frühzeitig an eine Lungentransplantation gedacht werden.

Autor:
OA Ass. Prof. Dr. Dr. Klaus Hackner
OA Ass. Prof. Dr. Dr. Klaus Hackner
Abteilung für Pneumologie, Universitätsklinikum Krems
Karl Landsteiner Privatuniversität für Gesundheitswissenschaften, Krems

Autor:
Dr. Mathis Hochrainer
Dr. Mathis Hochrainer
Abteilung für Innere Medizin und Pneumologie, Klinik Floridsdorf, Wien

Ursprünglich erschienen:
AEK 24|2023
AEK 24|2023
WAS PATIENT:INNEN WISSEN WOLLEN
Kann man Lungenfibrose heilen?
Eine klassische Lungenfibrose geht mit einer Vernarbung der Lunge einher, die nicht mehr rückgängig gemacht werden kann. Es gibt jedoch antifibrotische Medikamente, die den Krankheitsprozess bremsen bzw. manchmal sogar stoppen können.
Ist Lungenfibrose vererbbar?
Es gibt vor allem bei der idiopathischen Lungenfibrose (IPF) Erkrankungsfälle, die durch genetische Veränderungen ausgelöst werden, und damit auch Patient:innen aus Familien mit einer erhöhten Häufigkeit von Lungenfibrosen (bis zu 10 % aller IPF-Fälle). Vererbbare Lungenfibrosen werden derzeit intensiv untersucht. Es gibt aber auch weitere wichtige Risikofaktoren für Lungenfibrose, wie z. B. Tabakrauch.
Literatur bei den Verfassern
Bildnachweis
Vorschaubild: AdobeStock_566540069_Marcela Ruty Romero