


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hereditäres Angioödem
29. Mai 2020
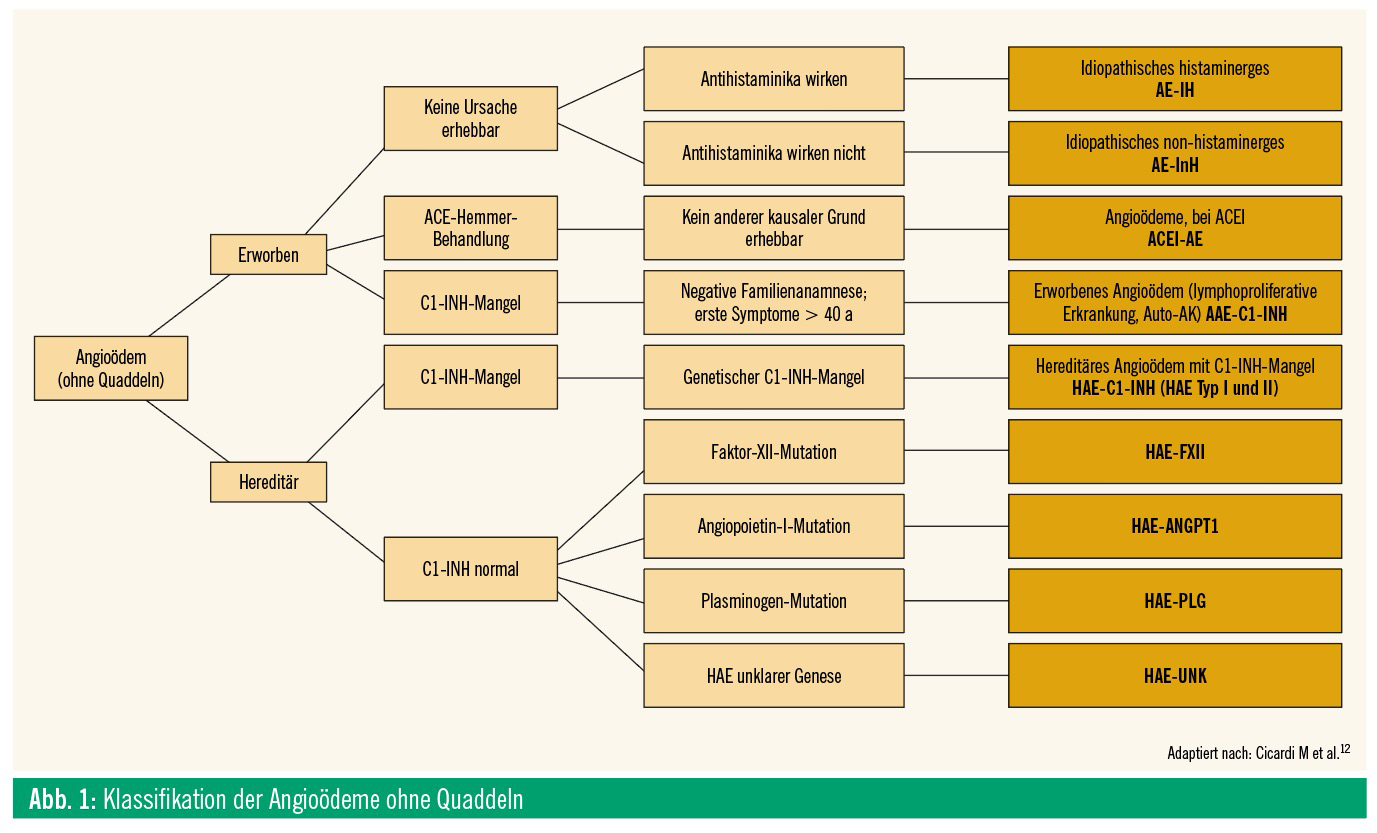
Beim hereditären Angioödem (HAE) handelt es sich um eine seltene, autosomal-dominant vererbte Erkrankung mit verschiedenen bisher beschriebenen Mutationen, welche endstreckig zu einer lokalen Erhöhung des Bradykininspiegels und somit zu einer Schwellung führen. Bisher sind Mutationen in mehreren Genen bekannt, die auch die Einteilung und Namensgebung bedingen (Abb. 1). Neben dem häufigeren HAE-C1-INH (erniedrigte C1-Inhibitor-Werte) gibt es die HAE-nC1- INH-Formen, welche mit einer normalen C1-INH-Funktion einhergehen (HAE-FXII, HAE-ANGPTI, HAE-PLG, HAE-UNK). Nicht zu vergessen ist auch das erworbene Angioödem (AAE-C1-INH), das beispielsweise im Rahmen von Lymphomen oder benignen monoklonalen Gammopathien (MGUS) auftreten kann.1 Das hereditäre Angioödem ist vom bradykininvermittelten, ACE-Hemmer-induzierten Angioödem (ACEI-AE) zu unterscheiden, das bei etwas weniger als 0,5 % der mit ACE-Hemmern behandelten Patienten auftritt. Individuelle, molekulargenetische Ursachen für das Auftreten von ACEI-AE sind im Wesentlichen unbekannt und Ziel aktueller Forschungsvorhaben.2
Charakteristisch für das hereditäre Angioödem (HAE-C1-INH) ist ein konstant erniedrigter Spiegel an funktionellem C1-INH, welcher periodisch und unvorhersehbar zu HAE-Schwellungsattacken führt. Dieser Mangel führt über eine unkontrollierte Aktivierung des Plasmakontaktsystems zu einer übermäßigen Bildung des Nonapeptids Bradykinin. C1-INH fungiert als endogener Kallikreininhibitor und verzögert so die Kininsynthese. Bei einer HAE-Attacke kommt es zur vorübergehenden Zunahme der lokalen Gefäßpermeabilität und zur Vasodilatation in den tiefen Schichten der Haut sowie der Schleimhäute. Die resultierende Schwellung bedingt das klinische Bild des hereditären Angioödems.3
Fehlender Juckreiz als Unterscheidungsmerkmal: Die rezidivierenden Ödeme sind in der Regel allerdings nicht von Juckreiz begleitet (wichtige Unterscheidung zu histaminvermittelten allergischen Angioödemen) und können jede Körperregion betreffen. Die Haut der Extremitäten (46,1 %), der Gastrointestinaltrakt (58,0 %), der Respirationstrakt sowie der urogenitale Bereich sind häufig – allein, aber auch in Kombination – von schmerzhaften Schwellungsattacken betroffen.4
Treten diese Attacken im Bereich der oberen Atemwege auf, können sie zum Tod durch Ersticken führen, sofern nicht rechtzeitig und mit geeigneten Medikamenten und/oder therapeutischen Interventionen (Sicherung der Atemwege/ Koniotomiebereitschaft) gegengesteuert wird. In Familien mit HAE war – und ist auch heute noch – der Erstickungstod bei Affektion der oberen Atem-Schluckstraße ein nicht seltenes, tragisches Ereignis. Eine retrospektive Auswertung zeigte, dass die Lebensdauer von erstickten Patienten mit undiagnostiziertem HAE-C1-INH im Durchschnitt etwa 31 Jahre kürzer ist als bei Patienten mit undiagnostiziertem HAE-C1-INH, die an anderen Ursachen starben. Vom Beginn der ersten Symptome bis zur Asphyxie vergehen oft nur wenige Stunden, in denen eine Behandlung lebensrettend sein kann.5, 6
Der „typische“ HAE-Patient
Üblicherweise präsentieren sich undiagnostizierte HAE-Patienten mit wiederholten Schwellungsattacken, die mehr als 24 Stunden andauern (bis zu 3–5 Tage) und nicht jucken. Bei der Inspektion des Patienten ist es wichtig, auf Quaddeln, Juckreiz oder Erytheme zu achten, da diese in der Regel beim HAE nicht vorkommen, sondern in erster Linie an eine histaminvermittelte Genese denken lassen. Für das Krankheitsbild ist aber das Erythema marginatum diagnostisch: ein gyriertes, teils flüchtiges Erythem, das der HAE-Attacke vorausgehen kann und von einem Teil der HAE-Patienten beschrieben wird, jedoch nicht an derselben Stelle auftreten muss, die von der Schwellungsattacke betroffen ist.
Rund 80 % der Patienten weisen eine positive Familienanamnese auf.4, 7 Das klinische Erscheinungsbild kann jedoch selbst innerhalb einer Familie, das heißt bei derselben Mutation, hinsichtlich Frequenz, Lokalisation und Stärke der Attacken stark variieren.
Präsentiert sich der Patient mit immer wieder auftretenden, kolikartigen Bauchschmerzen, die teils mit Diarrhö, Übelkeit und Erbrechen einhergehen, spricht man von abdominellen Attacken. Dabei kommt es zu einer sehr ausgeprägten Schmerzhaftigkeit, die unbehandelt mehrere Tage anhalten kann, bis sich die Schwellung wieder zurückbildet. Bei abdominellen Attacken ist oft im Gegensatz zu Hautattacken äußerlich keine Schwellung erkennbar, und bei der Ursachensuche stehen Differenzialdiagnosen wie Appendizitis, Cholezystitis, Pankreatitis etc. im Vordergrund. Kurz: HAE-Attacken im Gastrointestinaltrakt können sich vielgestaltig präsentieren, mitunter auch als Invagination des Dickdarms, wie ein Fallbericht zeigt; ein Notfall, der mittels Gabe von C1-INH-Konzentrat behandelt werden konnte.8
Bei Verdacht auf ein Angioödem kann die Schwellung der Darmwand mittels bildgebender Verfahren (CT, Ultraschall) dargestellt werden. Dabei zeigen sich typischerweise erweiterte Darmschlingen, Kokarden, verdickte Schleimhautfalten, aber auch – je nach Lokalisation – Aszites und freie Flüssigkeit im Douglas-Raum.9
Zu den bekannten häufigsten Triggern zählen mechanische Traumata, Stress und Atemwegsinfektionen (Abb. 2). Ein Großteil der Attacken tritt allerdings ohne erkennbare Trigger auf.

Diagnostische Methoden
Laborchemisch sind die Bestimmung des Komplementfaktors C4 und des funktionellen C1-INH-Wertes wegweisend. Beide Werte sind in der Regel erniedrigt, auch außerhalb der akuten Schwellungsattacken, wobei C4 mitunter im Normalbereich liegen kann und daher zum Ausschluss eines HAE-C1-INH auf jeden Fall der funktionelle C1-INH-Wert bestimmt werden muss.10 Die Bestimmung des C1-INH-Proteinspiegels dient lediglich zur Differenzierung zwischen HAE-C1-INH Typ I (HAE-1, mit verminderter C1-INH-Funktionalität und verminderten C1-INH-Spiegeln) und Typ II (HAE-2, mit verminderter C1-INH-Funktionalität und erhöhten/ normalen C1-INH-Spiegeln); klinisch und therapeutisch ist die Unterscheidung jedoch unwesentlich.
Eigene Untersuchungen (noch nicht publiziert) zeigen, dass der funktionelle C1-INH-Wert ein sehr zeit-, temperatur- und aufarbeitungssensibler Laborparameter ist, weswegen nach der Probennahme eine rasche und exakte Probenuntersuchung wesentlich ist. Molekulargenetische Analysen der entsprechenden Gene sind zwar einerseits beweisend für das Krankheitsbild, im typischen Fall für die Diagnosefindung aber nicht zwingend erforderlich.
Eine rezente Publikation zum Thema HAE in Österreich identifizierte 137 HAE-C1-INH-Patienten, wobei beträchtliche lokale Unterschiede hinsichtlich der Patientenzahl in den Bundesländern auffallend sind. Männer und Frauen sind gleich häufig betroffen. Die Prävalenz beträgt in Österreich in etwa 1 : 64.000. Die Studie zeigt, dass das mediane Alter bei Auftreten der ersten Symptome 6,5 Jahre beträgt, wobei der Großteil der Betroffenen die ersten Symptome in den ersten beiden Lebensjahrzehnten berichtet. Der Blick auf das mediane Alter bei Diagnosestellung von 21,0 Jahren offenbart eine enorme diagnostische Verzögerung, die sich erst bei der jüngeren Generation der Betroffenen reduziert.7
Therapie
Aktuell stehen in Österreich mehrere Optionen für Akuttherapie, Kurzzeit- und Langzeitprophylaxe zur Verfügung. Bis vor einigen Jahren waren in den meisten Ländern nur attenuierte Androgene, Antifibrinolytika und gefrorenes Frischplasma (FFP) für die Behandlung des HAE vorhanden; die Wirkkraft dieser Mittel war unzuverlässig und ihr Nebenwirkungsprofil problematisch. Bis heute existiert keine kurative Therapie. Zwei Plasmaprodukte (Berinert® und Cinryze®) sowie ein rekombinant hergestellter rC1-INH (Ruconest®) stehen als intravenöse C1-INH-Substitutionspräparate zur Verfügung. Daneben kann auch der Bradykinin-B2-Rezeptor-Antagonist Icatibant subkutan zur Akutbehandlung eingesetzt werden. Die frühzeitige Therapie der Attacke ist wesentlich, da durch sie lediglich das Fortschreiten der Schwellung gestoppt wird; auf die passive Resorption des Extravasats haben die eingesetzten Therapeutika jedoch keinen Einfluss. Seit 2019 ist der Kallikrein-Inhibitor Takhzyro® als subkutanes Prophylaktikum verfügbar. Demnächst wird auch die subkutane C1-INH-Prophylaxe für jugendliche und erwachsene HAE-Patienten (Typ 1 und 2) zur Verfügung stehen. An oralen Kallikrein-Inhibitoren zur Akutbehandlung und für die Prophylaxe wird intensiv geforscht.11
Resümee
Es sind in Österreich 137 HAE-C1-INH-Patienten dokumentiert; die Zahl undiagnostizierter Betroffener dürfte aber ähnlich hoch sein. Die diagnostische Verzögerung ist nach wie vor unverhältnismäßig lang. Es handelt sich bei den HAE-Attacken um bradykininvermittelte Ödeme; eine antiallergische Therapie mit Antihistaminika und Kortikosteroiden ist daher wirkungslos.
Interessenskonflikt: Clemens Schöffl: Referent für Shire, Unterstützung für Kongressreisen von Shire, CSL Behring und Biocryst; klinische Studien mit Shire und Biocryst.
Werner Aberer: Teilnehmer an Advisory Boards und Vorträge für Biocryst, CSL Behring, Pharming und Shire; klinische Studien mit Biocryst und Shire.
1 Maurer M et al., Allergy 2018; 73:1575–96
2 Sachs B et al., Hautarzt 2018; 69:298–305
3 Nussberger J et al., Lancet 1998; 351: 1693–7
4 Banerji A et al., Ann Allergy Asthma Immunol 2020; [Online ahead of print]
5 Arnoldsson H et al., Acta Med Scand 1967; 181:115–24
6 Bork K et al., J Allergy Clin Immunol 2012; 130:692–7
7 Schöffl C et al., J Dtsch Dermatol Ges 2019; 17:416–23
8 Patel N et al., Case Reports Immunol 2015; 2015:925861
9 Dirks K et al., Ultraschall Med 2001; 22:186–90
10 Tarzi MD et al., Clin Exp Immunol 2007; 149:513–6
11 Aygoren-Pursun E et al., N Engl J Med 2018; 379:352–62
12 Cicardi M et al., Allergy 2014; 69:602–16
13 Caballero T et al., J Investig Allergol Clin Immunol 2016; 26:383–86

AutorIn: Dr. Clemens Schöffl
Universitätsklinik für Dermatologie und Venerologie, Medizinische Universität Graz
Foto: Werner Stieber

AutorIn: Univ.-Prof. Dr. Werner Aberer
Universitätsklinik für Dermatologie und Venerologie, Medizinische Universität Graz
Foto: Medizinische Universität Wien/Felicitas Matern

Ursprünglich erschienen:
SD 02|2020
SD 02|2020
Herausgeber: Ao. Univ.-Prof. Dr. Christoph Höller, Assoc. Prof. Priv.-Doz. Dr. Constanze Jonak, Ao. Univ.-Prof. Dr. Rainer Kunstfeld, Universitätsklinikfür Dermatologie, Wien; Univ.-Prof. Dr. Hubert Pehamberger, Wien
Publikationsdatum: 2020-05-28
Zur Ausgabe »
Publikationsdatum: 2020-05-28
Zur Ausgabe »
