


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Therapie der Hyperlipidämie – Vergangenheit, Gegenwart und Zukunft
26. April 2019
Hyperlipidämien stellen einen Risikofaktor für die Entwicklung von kardiovaskulären und koronaren Gefäßerkrankungen dar. Die zunehmende Anzahl von Übergewichtigen und Fettleibigen führt zu immer mehr Patienten mit Störungen im Fettstoffwechsel. Über 50 % der Erwachsenen und über 15 % der Kinder aus OECD-Staaten waren 2017 übergewichtig, Tendenz bis 2030 steigend.1
Die Hypercholesterinämie ist die häufigste Dyslipidämie. Sie ist durch erhöhte Plasmacholesterin- und Lipoproteinspiegel charakterisiert. Lipoproteine sind Komplexe aus Apolipoproteinen und Lipiden. Sie ermöglichen den Transport der nichtwasserlöslichen Lipide und Vitamine im Blut. Das Low-Density Lipoprotein (LDL), High Density Lipoprotein (HDL) und Lipoprotein (a) (Lp[a]) stellen die bekanntesten Vertreter dar. Die Bedeutung des Cholesterins in der Entstehung der Atherosklerose und entsprechende Therapien wurden im 20. Jahrhundert intensiv beforscht (Tab. 1).
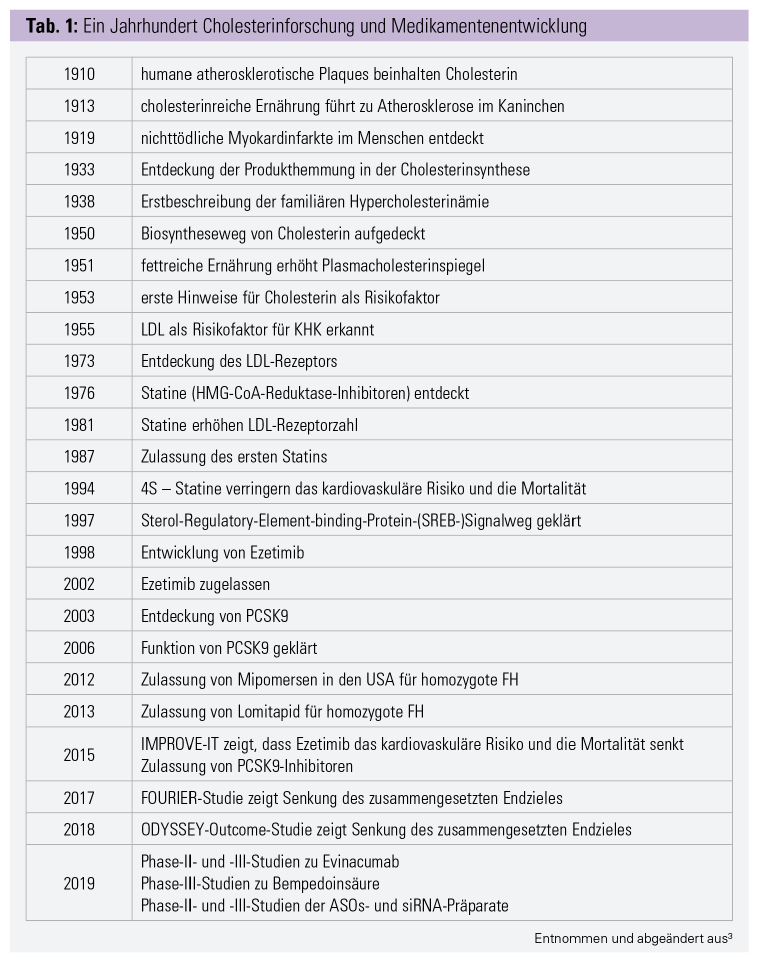
Die vergangenen 3 Jahrzehnte
Statine: 1987 wurde von der U. S. Food and Drug Administration (FDA) das erste Statin zugelassen. Statine hemmen die 3-Hydroxy-3-Methylglutaryl-Coenzym-A-Reduktase (HMG-CoA-Reduktase).2, 3 Die entsprechende enzymatische Reaktion ist der geschwindigkeitsbestimmende Schritt in der Cholesterinsynthese in der Leber.3–5 Die Hemmung der Synthese führt zu einer kompensatorischen Erhöhung der LDL-Rezeptorzahl, welche das an LDL gebundene Cholesterin aus der Zirkulation entfernt. Statininduzierte Myopathien werden häufig als Nebenwirkung gemeldet. Die wahre Inzidenz dafür liegt lediglich bei 1 : 10.000. Polymorphismen im SLCO1B1-Gen – dem leberspezifischen Statintransporter – erhöhen die Plasmakonzentration der Statine und damit die Wahrscheinlichkeit für Nebenwirkungen.6
Die Scandinavian Simvastatin Survival Study (4S) zeigte 1994 noch nie erzielte Ergebnisse. Simvastatin senkte das LDL-Cholesterin (LDL C) um 35 %, das relative Risiko für kardiovaskuläre Tode um 42 %. Zudem reduzierte es das relative Risiko für schwere kardiovaskuläre Ereignisse, für koronare Ereignisse, für Bypässe und für Interventionen. 4S war die erste Studie, die innerhalb des geplanten Beobachtungszeitraums eine signifikante Senkung der Gesamt- und kardiovaskulären Mortalität durch LDL-C-Senkung beweisen konnte.7 Hochpotente Statine – Rosuvastatin und Atorvastatin – senken das LDL-C um bis zu 50 % und sind heute die Mittel der Wahl.8
2002 wurde Ezetimib von der FDA zugelassen. Es hemmt die Aufnahme von biliärem- und Nahrungscholesterin am Bürstensaum der Dünndarmenterozyten. Es wirkt über das Niemann-Pick-C1-like-Protein – ein Steroltransporter, welcher durch Ezetimib inhibiert wird.9 Die Kombination von Simvastatin mit Ezetimib senkt das LDL-C um zusätzliche 24 %. 2015 belegte die IMPROVE-IT-Studie, dass Ezetimib in Kombination mit einem Statin das Risiko für den zusammengesetzten Endpunkt signifikant senkt.10
PCSK9-Hemmung: 2015 wurden Alirocumab und Evolocumab zugelassen. Beide Wirkstoffe sind monoklonale Antikörper gegen die Proproteinkonvertase Subtilisin/Kexin Typ 9 (PCSK9). PCSK9 sorgt im Gesunden dafür, dass der LDL-Rezeptor nach der Bindung und Endozytose abgebaut wird. Durch die Inhibition wird der Rezeptor nicht abgebaut und rezirkuliert – bis zu über hundertmal. Die Rezeptorgesamtzahl nimmt zu, und es wird mehr LDL-C aus dem Kreislauf aufgenommen.
ODYSSEY Outcomes (Alirocumab) und FOURIER (Evolocumab) – zwei große Cardiovascular Outcome Trials (CVOT) – zeigten, dass PCSK9-Inhibitoren, zusätzlich zu einer bestehenden hochpotenten Statintherapie, eine weitere LDL-C-Senkung um etwa 50 % ermöglichen. PCSK9-Inhibitoren senken das Risiko für koronare und kardiovaskuläre Ereignisse, für Myokardinfarkte, Schlaganfälle und koronare Revaskularisationen. Diese Medikamentenklasse stellt damit eine Therapieoption für statinintolerante und therapieresistente Patienten dar.11, 12
Lomitapid: Die Produktion des Hauptbestandteils von LDL – Apolipoprotein B – kann durch Lomitapid inhibiert werden. Dadurch kann das LDL-C um 50 % gesenkt werden. Bei Einnahme kommt es jedoch zu Erhöhungen der Leberenzyme und zu Fetteinlagerungen in der Leber. Daher hat Lomitapid lediglich eine Zulassung für die homozygote familiäre Hypercholesterinämie. Durch das bedenkliche Nebenwirkungsprofil, die hohen Kosten und die eingeschränkte Indikation ist die Anwendung stark limitiert.13–17
Was die Zukunft bringt
Der Blick in die Zukunft verspricht einige neue interessante Therapien (Tab. 2). Derzeit beginnen mehrere Phase-III-Studien mit dem Wirkstoff Bempedoinsäure. Sie hemmt die Cholesterinsynthese und erhöht die LDL-Rezeptorzahl, indem sie die ATP-Citrat-Lyase – ein Enzym der Cholesterinsynthese – inhibiert. Zusätzlich zu Ezetimib senkt Bempedoinsäure das LDL-C bei Patienten mit Statinunverträglichkeit um 23,5 %.18 Der Wirkstoff wird nur in der Leber aktiviert, im Muskel kommt das entsprechende Enzym nicht vor, das soll die muskulären Nebenwirkungen verhindern.19, 20
Eine Loss-of-Function-(LOF-)Mutation im Angiopoietin-like 3-(ANGPTL3-)Gen zeigte erniedrigte LDL-C-Werte und Triglyzeride (TG) im Tier und in genomweiten Assoziationsstudien.21, 22 Ein dementsprechend wirkender monoklonaler Antikörper – Evinacumab – befindet sich in Phase II und III. Er zeigte im Tier und im Menschen vielversprechende Effekte.23
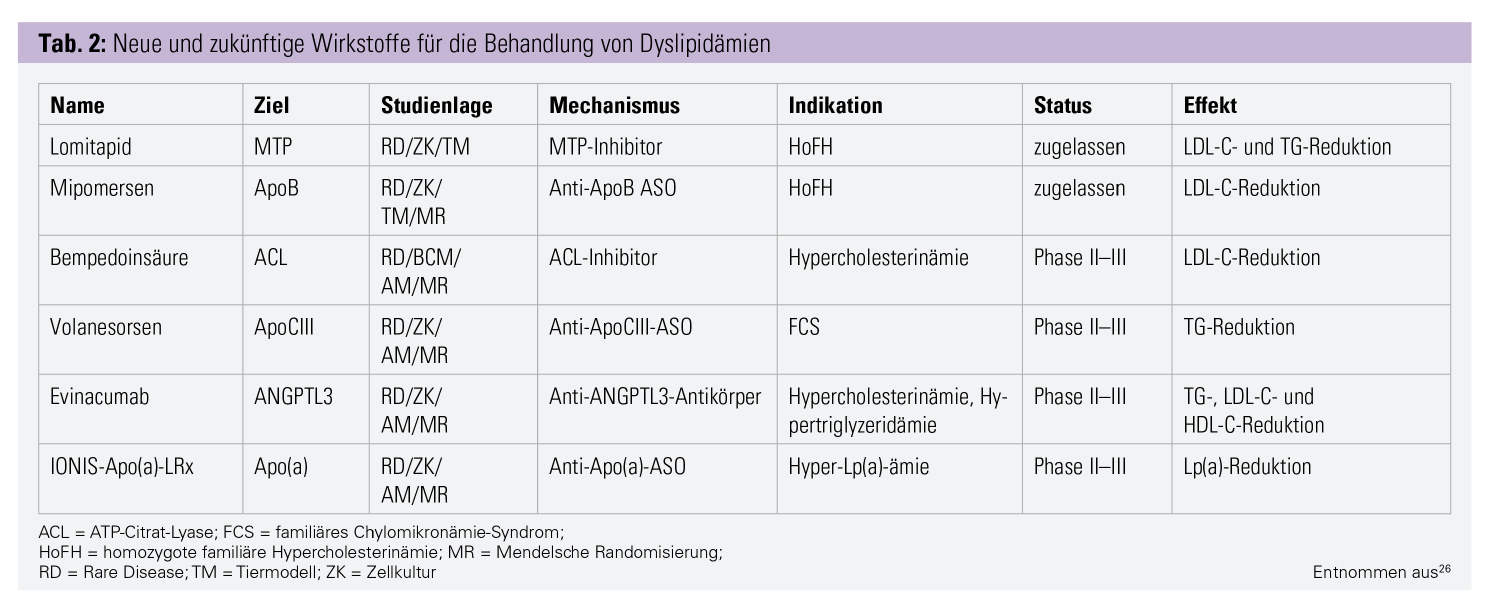
Antisense-Oligonukleotide und siRNA: Genetische Mutationen sind aus der Entwicklung von Pharmaka nicht mehr wegzudenken. Sie helfen beim Entdecken neuer Targets und ermöglichten es, neue Wirkmechanismen zu entwickeln. Das in Österreich nicht erhältliche Mipomersen war das erste Antisense-Oligonukleotide (ASO) in der Behandlung von Hyperlipidämien. ASOs sind modifizierte einzelsträngige DNA- oder RNA-Moleküle. Sie binden und deaktivieren hochspezifisch die mRNA des Zielproteins in der Zelle. siRNA-Präparate (Small interfering RNA) wirken sehr ähnlich, jedoch handelt es sich dabei um doppelsträngige Moleküle.17 Ein siRNA-Präparat gegen PCSK9, welches LDL-C um bis zu 50 % senken soll, befindet sich momentan in der Phase III, es heißt Inclisiran. Ein weiteres ASO hat die mRNA von Apolipoprotein C3 (ApoC3) als Ziel. LOF-Mutationen im ApoC3-Gen führen zu niedrigeren Triglyzerid-(TG-)Spiegeln und reduzieren so das Risiko für koronare und kardiovaskuläre Erkrankungen. Mit diesem Wirkmechanismus senkt Volanesorsen TG um 71 %. Es befindet sich momentan in Phase III.17, 24 Lp(a) ist ein unabhängiger Risikofaktor für kardiovaskuläre Erkrankungen, über dessen Funktion diskutiert wird; rund 20 % der Bevölkerung haben Lp(a)-Erhöhungen.25 Momentan befindet sich ein ASO in der Phase II, die Phase III läuft 2020 an. Dieses Präparat senkt Lp(a) um 92 %. Dies wäre die erste spezifische Therapie für die direkte Senkung von Lp(a).17
Resümee
Lipidologische Therapien befinden sich im Umbruch und die nächsten Jahre versprechen interessante und neue Therapieansätze für die Behandlung von Hyperlipidämien. Für bisher kaum therapierbare Hyperlipidämien, wie die Hyper-Lp(a)-ämie und die Hypertriglyzeridämie, dürften in baldiger Zukunft neue Therapieoptionen verfügbar sein.
1 OECD. OECD OBESITY UPDATE 2017: OECD; 2017 [cited 28. 08. 2018]. Available from: http://www.oecd.org/health/obesity-update.htm
2 Endo A et al., FEBS letters 1976; 72(2):323–6
3 Goldstein JL et al., Cell 2015; 161(1):161–72
4 Zetterstrom R, Acta paediatrica 2009; 98(7):1223–7
5 Bloch K, Science 1965; 150(3692):19–28
6 Group SC et al., N Engl J Med 2008; 359(8):789–9
7 ScandinavianSimvastatinSurvivalStudyGroup, Lancet 1994; 344(8934):1383–9
8 Stalenhoef AF et al., Eur Heart J 2005; 26(24):2664–72
9 Garcia-Calvo M et al., Proc Natl Acad Sci USA 2005; 102(23):8132–7
10 Cannon CP et al., N Engl J Med 2015; 372(25):2387–97
11 Sabatine MS et al., N Engl J Med 2017; 376(18):1713–22
12 Schwartz GG et al., N Engl J Med 2018; 379(22):2097–107
13 Cuchel M et al., N Engl J Med 2007; 356(2):148–56
14 Cuchel M et al., Lancet 2013; 381(9860):40–6
15 Raal FJ et al., Lancet 2010; 375(9719):998–1006
16 Santos RD et al., Eur Heart J 2015; 36(9):566–75
17 Tsimikas S, Curr Opin Lipidol 2018; 29(6):459–66
18 Ballantyne CM et al., Atherosclerosis 2018; 277:195–203
19 Bilen O et al., Curr Atheroscler Rep 2016; 18(10):61
20 Pinkosky SL et al., Nat Commun 2016; 7:13457
21 Koishi R et al., Nat Genet 2002; 30(2):151–7
22 Willer CJ et al., Nat Genet 2008; 40(2):161–9
23 Dewey FE et al., N Engl J Med 2017; 377(3):211–21
24 Jorgensen AB et al., N Engl J Med 2014; 371(1):32–41
25 Gencer B et al., Eur Heart J 2017; 38(20):1553–60
26 Hegele RA et al., Circ Res 2019; 124(3):386–404

AutorIn: Ao. Univ.-Prof. Dr. Christoph Ebenbichler
Schwerpunkte: Gastroenterologie, Hepatologie, Endokrinologie und Stoffwechsel, Universitätsklinik für Innere Medizin I, Medizinische Universität Innsbruck

Ursprünglich erschienen:
UIM 03|2019 Themenheft Kardiologie
UIM 03|2019 Themenheft Kardiologie