


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
INITIATIVE BLUTGEFÄSSE VERSTEHEN: Morbus Horton – Kopfschmerzen führen zur Erblindung
12. November 2012
Nachdem die Inzidenz mit zunehmendem Alter weiter ansteigt, wird mit dem Überaltern unserer Bevölkerung auch eine zunehmende Häufung der Arteriitis temporalis beobachtet. Die wissenschaftliche Erstbeschreibung erfolgte 1932 durch Horton, Magath und Brown. Durch deren Fallbericht einer temporalen Arteritis, welche mit Erblindung, Zungennekrose und Kieferclaudicatio einherging, bekam diese Erkrankung schließlich die Bezeichnung „Morbus Horton“ (M. Horton). Die Inzidenz der Arteriitis cranialis beträgt im europäischen Raum ca. 20 Fälle pro 100.000 pro Jahr, Frauen sind ca. dreimal häufiger betroffen.
Ursache und Pathomechanismus
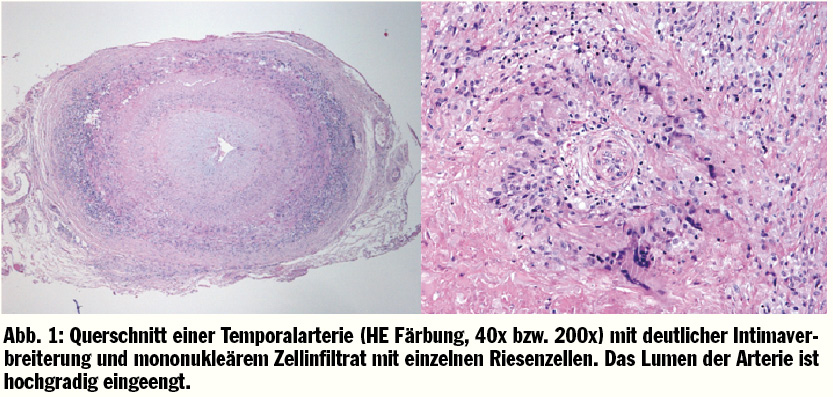
Die Ursache dieser entzündlich bedingten Gefäßerkrankung ist noch weitgehend unbekannt. Es werden verschiedene auslösende Faktoren diskutiert. Bekannt ist, dass es eine T-Zell-vermittelnde autoimmunologische Ursache mit genetischer Disposition gibt. Zusätzlich werden stattgehabte Infektionen als weiterer ursächlicher Faktor diskutiert. Im Bereich der betroffenen Gefäßabschnitte werden mononukleäre Zellinfiltrate aus Makrophagen und CD4 positiven Zellen nachgewiesen, welche eine antigenspezifische T-Zellreaktion auslösen. Diese Infiltrate führen zu granulomatösen Zellansammlungen, den sogenannten Riesenzellen. In weiterer Folge führt ein Gewebsumbau mit hyperplastischen Myofibroblasten schließlich zu einer Stenose des Gefäßlumens (Abb. 1). Diese Gefäßveränderungen sind durch einen segmentalen Befall gekennzeichnet. Die beschriebenen granulomatösen Veränderungen werden in den Aortenästen 2.–5. Ordnung mit einer bevorzugten Lokalisation in den Aa. temporales, den Aa. ciliares posteriores, den Aa. vertebrales, den Aa. ophthalamicae, wie auch in den Ästen der A. carotis externa beobachtet. Die entzündlich bedingte Stenose kann schließlich zu einem vollständigen Verschluss des Gefäßes mit entsprechender Ischämie-Symptomatik des jeweiligen arteriellen Stromgebietes führen.
Klinik
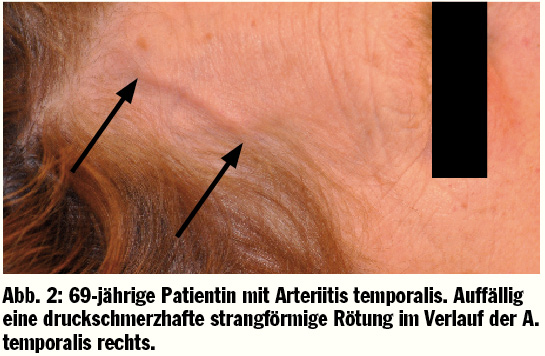
Die Klinik des M. Horton beginnt meist unspezifisch. Die Patienten leiden an Myalgien und klagen über Schwäche, Abgeschlagenheit und ein deutliches Krankheitsgefühl. Zusätzlich wird ähnlich zu verschiedenen malignen Erkrankungen häufig eine sogenannte „B-Symptomatik“ mit subfebrilen Temperaturen, Gewichtsabnahme und nächtlichem Schwitzen beobachtet. Das in dreiviertel aller Fälle einer Arteritis cranialis vorliegende Hauptsymptom sind starke, pulsierende temporal lokalisierte Kopfschmerzen, welche meist ein- oder auch beidseitig auftreten. Zusätzlich werden gelegentlich oberflächlich empfundene Schmerzen der Kopfhaut beschrieben. Ein weiteres Symptom, welches bei ungefähr 40% der Patienten beobachtet wird, ist die Claudicatio der Kau-, Schlund- und Zungenmuskulatur, die sogenannte Kieferclaudicatio. Die Patienten klagen über starke Schmerzen beim Kauen, wobei der Schmerz im Gegensatz zur gelenksbezogenen Kiefergelenksarthrose im Bereich der Muskulatur verspürt wird. Die kopfbezogenen Beschwerden lassen sich durch den entzündlichen Befall der extrakraniellen Kopfarterien, sowie die entzündlichen Veränderungen der A. temporalis erklären. Im Bereich der A. temporalis sind knotige Verhärtungen mit einer starken Berührungsempfindlichkeit auffällig (Abb. 2).
Eine entzündliche Beteiligung der Ziliararterien mit konsekutiver ischämischer Optikusneuropathie kann zu einer akut einsetzenden Visuseinschränkung führen. Durch diese okuläre Beteiligung kann es ohne unmittelbare Behandlung innerhalb weniger Tage zu einem vollständigen Visusverlust kommen. Die Wahrscheinlichkeit einer okulären Beteiligung ist mit 50% sehr hoch, wobei die Visusbeeinträchtigung im Rahmen der ischämischen Optikusneuropathie zumeist irreversibel ist. Aus diesem Grunde ist auch die rechtzeitige Diagnostik und Therapie des M. Horton essenziell.
Diagnostik
Da es weder spezifische klinische Symptome noch Laborparameter zur Diagnosestellung der Arteriitis temporalis gibt, besteht der Diagnoseprozess in einer Beurteilung von Klinik, Laborveränderungen, Bildgebung der großen Arterien und schließlich Biopsie der Temporalarterie. Die 1990 publizierten Kriterien des American College of Rheumatology (ACR) sind nach wie vor gültig für die Klassifikation der Riesenzellarteriitis (Tab.). Bei Vorliegen von ≥ drei der fünf Kriterien kann eine Sensitivität von 90,5% und Spezifität von 97,8% in der Differenzierung gegenüber anderen Vaskulitiden angenommen werden.
In der Praxis sollte vor allem bei Patienten mit einem Alter über 50 Jahre, welche wegen akut aufgetretenen Kopfschmerzen, Schmerzen der Kopfhaut, Kieferclaudicatio, Sehstörungen und/oder deutlicher Verschlechterung des Allgemeinbefindens vorstellig werden, an eine mögliche Riesenzellarteriitis gedacht werden. Die Blutsenkungsgeschwindigkeit (BSG) ist der am häufigsten verwendete Laborparameter um diese Verdachtsdiagnose zu unterstützen. Gemäß den ACR-Kriterien wird eine BSG von ≥50 mm/h als positives Kriterium gewertet, wobei Patienten mit einer Arteriitis temporalis häufig Werte jenseits von 80 mm/h aufweisen. Weitere mögliche Laborveränderungen sind neben einer Erhöhung des C-reaktiven Proteins (CRP) eine mikrozytäre Anämie, Thrombozytose, eine erhöhte Alkalische Phosphatase, oder erhöhte Alpha-1 und Alpha-2 Globuline der Serumelektrophorese.

Der klinische Verdacht einer Riesenzellarteriitis sollte schließlich zu einer umgehenden Abklärung mittels Duplexsonographie der Temporalarterien und supraaortalen Gefäße einschließlich der Vertebralarterien führen. Ein entzündlich bedingtes Ödem der arteriellen Gefäßwand erscheint als hypoechogener Halo um die betroffenen Gefäße (Sensitivität 69%, Spezifität 82%). Zusätzlich können die entzündlichen Veränderungen zu Stenosierungen bis hin zu einem hypoechogenen Verschluss führen. Der Goldstandard der Diagnostik besteht in der Biopsie der Temporalarterie, wobei aufgrund möglicher skip-lesions ein ausreichend langes Stück (mind. 2 cm) bevorzugt an der symptomatischen Seite entnommen werden sollte. Eine unmittelbare Therapie sollte bei klinischem Verdacht auf RZA jedoch aufgrund der möglichen irreversiblen Visusbeeinträchtigung unabhängig vom Zeitpunkt der Biopsie stets angestrebt werden.
Therapie und Verlauf
Das Ziel der Therapie besteht darin ein mögliches Fortschreiten von Sehveränderungen bzw. eine Beteiligung des vielleicht noch nicht betroffenen Auges zu verhindern, sowie die restlichen klinischen Symptome (Kopfschmerzen, Kieferclaudicatio, Gewichtsverlust etc.) zum Sistieren zu bringen. Eine systemische Corticoidtherapie ist die Basis in der Therapie der Arteriitis temporalis. Es gibt keine einheitliche Therapieempfehlung bezüglich der Cortisondosierung. In Anlehnung an die Richtlinien der Europäischen Fachgesellschaft für Rheumatologie (EULAR-Recommendations) kann eine Dosierung von 1mg/kg Methylprednislon (max. 60 mg/Tag) empfohlen werden. Diese Dosis sollte für einen Monat beibehalten werden und anschließend stufenweise reduziert werden. Bei bereits vorliegender Augenbeteiligung wird in einigen Zentren eine wesentlich höhere Initialdosierung (bis zu 1 g/Tag i.v. über drei Tage mit anschließender Dosisreduktion) empfohlen. Unabhängig von der Visusbeeinträchtigung wird bei vielen Patienten eine längerfristige Cortisontherapie durchgeführt. Um mögliche Steroid-induzierte Nebenwirkungen zu minimieren, wird in diesen Fällen eine begleitende immunsuppressive Therapie (z.B. mit 10–15 mg Methotrexat pro Woche) empfohlen. Zusätzlich zu dieser immunmodulierenden Therapie wird zur Prophylaxe zerebraler Ischämien eine Thrombozytenfunktionshemmung (z.B. Acetylsalicylsäure 100 mg/Tag) verabreicht.
Bis auf die meist irreversible Visusbeeinträchtigung bessert sich die Symptomatik in der Regel bereits innerhalb der ersten Tage nach Einleiten der Steroidtherapie. Dennoch ist aufgrund des großen Rezidivrisikos in den meisten Fällen eine Therapiedauer von zumindest ein bis zwei Jahren notwendig.
In seltenen Fällen muss die Therapie aufgrund eines chronisch relapsierenden Verlaufes über mehrere Jahre beibehalten werden. Aufgrund des unterschiedlichen Verlaufes ist eine entsprechende Nachsorge und individuelle Therapieentscheidung hinsichtlich einer möglichen Dosisreduktion der Cortisontherapie notwendig. Diese Entscheidung wird jeweils abhängig von der Klinik, der serologischen Entzündungsaktivität (BSG, CRP) und des bisherigen Verlaufes der Erkrankung getroffen. Durch dieses Therapieregime kann eine niedrige Relaps-Rate erreicht werden und eine sekundäres Erblinden bei einem Großteil der Patienten verhindert werden.

Ursprünglich erschienen:
AEK 21|2012
AEK 21|2012