


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Mycosis fungoides: das häufigste primär kutane T-Zell-Lymphom
2. Juni 2023
Die Mycosis fungoides (MF) ist ein Misnomer für eine eigenständige Entität innerhalb der primär kutanen T-Zell-Lymphome (CTCL). Der Terminus MF ist der bildlichen Namensgebung der Dermatologie des 19. Jahrhunderts geschuldet. Heute sind CTCL den Non-Hodgkin-Lymphomen (NHL) zugeordnet. Ätiologisch werden Infektionserreger, ultraviolettes Licht (UV) oder auch berufliche Exposition als mögliche Trigger kontrovers diskutiert. Zudem sind genomische Veränderungen in putativen Onkogenen und Tumorsuppressorgenen einschließlich NF-ƙB und des Janus-STAT-Signalweges belegt.
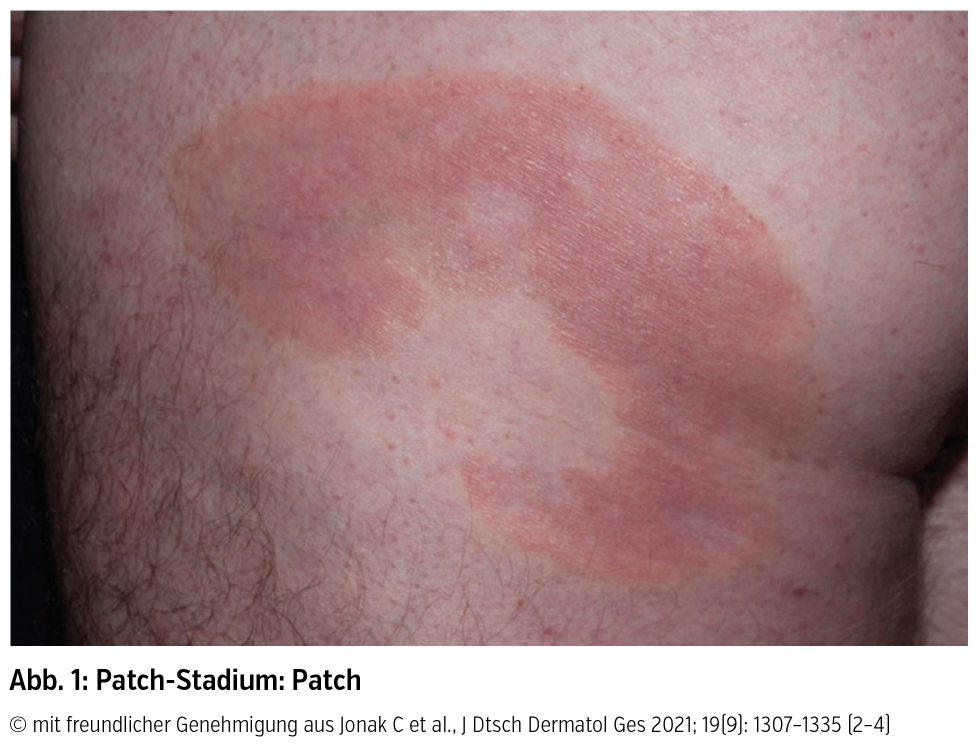
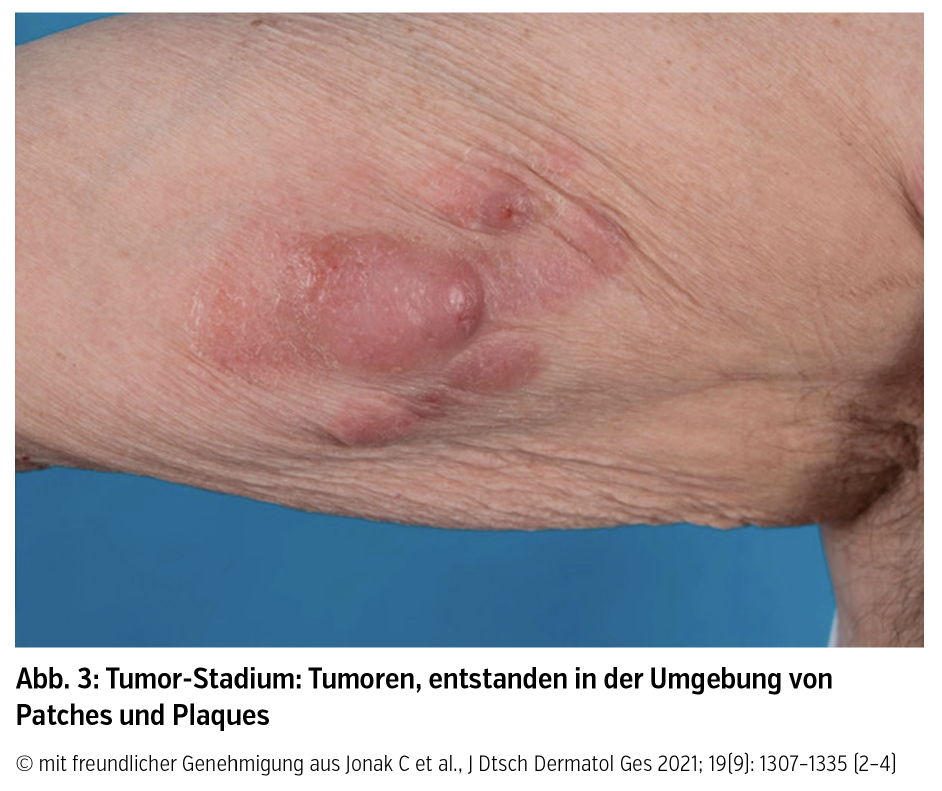
Klinik und Verlauf: Der Altersgipfel für die Diagnose liegt zwischen 55 und 60 Jahren (m : w = 2 : 1). Für die MF ist klinisch ein stadienhafter Verlauf von Patches über Plaques bis hin zu Tumoren dokumentiert (Abb. 1–3), sowie deren Koexistenz oder auch das Überspringen von einzelnen Stadien:
Patches sind als erythematöse Maculae mit variabler Desquamation und Größe definiert und finden sich lokalisiert oder generalisiert vor allem in sonnengeschützten Hautarealen. Obwohl sie als klinisches Merkmal für die frühe MF gelten, finden sich Patches auch bei fortgeschrittener Krankheit oder als Rezidiv nach erfolgreicher Therapie von Plaques oder Tumoren. Plaques sind hingegen als infiltrierte erythematös bis bräunliche Läsionen mit irregulärer Begrenzung und variabler epidermaler Komponente charakterisiert und müssen von flachen Tumoren abgegrenzt werden. Tumoren sind definiert durch einen Durchmesser ≥ 1 cm und entwickeln sich bei bestehenden Patches und Plaques oder fallweise de novo. Als viertes Hautstadium neben Patch-, Plaque- und Tumorstadium kann sich auch eine Erythrodermie (generalisierte Rötung der Haut) bei MF manifestieren.
 |
 |
Durch die Krebsdiagnose selbst sowie durch das sichtbare Stigma der Hautläsionen mit oft quälendem Pruritus ist die gesundheitsbezogene Lebensqualität (HRQoL) von MF-Patient:innen nachweislich beeinträchtigt.
Diagnose, Differenzialdiagnose, Prognose
Differenzialdiagnostisch muss die MF im Initialstadium von anderen Dermatosen wie beispielsweise einem mikrobiellen oder atopischen Ekzem, einer Psoriasis vulgaris und einer Pityriasis rosea (Röschenflechte) unterschieden werden. Zudem gestaltet sich die histopathologische Diagnose der MF trotz genauer Definition insbesondere im frühen Stadium der Erkrankung oft sehr schwierig und ist meist nur durch die genaue Korrelation mit der Klinik möglich. Somit ist es auch nachvollziehbar, dass die Diagnose der frühen MF verzögert im Mittel erst nach 36 Monaten gestellt wird.
Die Diagnostik basiert auf einer ausführlichen Anamnese, einem genau dokumentierten Hautbefund, histologischen Untersuchungen, Laboruntersuchungen (inklusive Durchflusszytometrie bzw. FACS), die in besonderen Fällen auch eine Immunphänotypisierung und/oder molekularbiologische Tests von Blut und Knochenmark beinhalten, sowie auf bildgebenden Verfahren, wie z. B. Lymphknotensonografie und Ganzkörper-CT oder PET-CT. Dabei muss unbedingt darauf geachtet werden, dass die molekularbiologischen und histologischen Befunde gegenüber der klinischen Präsentation und Anamnese nicht überbewertet werden, da ansonsten das Risiko einer Überbehandlung besteht. So liegt beispielsweise nicht selten auch bei entzündlichen Erkrankungen eine molekularbiologische Monoklonalität vor, deren Nachweis daher diagnostisch und prognostisch oft nur bedingt relevant ist.
Das Staging der MF basiert auf der revidierten, international anerkannten TNM-Klassifikation der International Society for Cutaneous Lymphomas (ISCL) und der European Organization for Research and Treatment of Cancer (EORTC). Diese beinhaltet neben der Hautbeteiligung – Primärtumor (T), Lymphknotenbefall (N) und Fernmetastasen (M) – auch den Nachweis von atypischen Lymphozyten bzw. Tumorzellen im peripheren Blut (B). Sie wird daher als „TNMB-Klassifikation“ bezeichnet. Anhand dieser Klassifikation erfolgt die Einteilung der Erkrankung in vier klinische Stadien. Jedes Stadium ist wiederum mit einem geschätzten Überleben (5-Jahres-Überlebensrate) assoziiert und somit aktuell bei bislang fehlenden prognostischen (Bio-)Markern prognostisch relevant.
Hinsichtlich der wenigen bekannten prognostischen Faktoren abseits des klinischen Stadiums erhob eine univariate retrospektive Analyse, dass hohes Alter, männliches Geschlecht, ein erhöhter Laktatdehydrogenase-Wert (LDH-Wert) und eine LCT das Überleben verschlechtern bzw. das Risiko für eine Progression der Erkrankung erhöhen. Rezente Daten bezüglich prognostisch geschlechtsbezogener Unterschiede bei MF ermittelten bei Frauen ein 5-Jahres-Gesamtüberleben von 76,9 % vs. 70,7 % bei Männern, wobei die Effekte des Östrogens auf die Differenzierung von Lymphomzellen und die Anti-Tumor-Immunantwort als ursächlich diskutiert werden.
Therapieoptionen
Die Behandlung der MF erfolgt basierend auf Leitlinien, in Europa überwiegend gemäß der EORTC-Konsensus-Therapieempfehlungen. In allen Leitlinien für die Behandlung von CTCL besteht Einigkeit, dass der Therapieansatz bei aktueller Unheilbarkeit (abgesehen von allogener Stammzelltransplantation) palliativ ist. Demnach sollen in frühen Stadien der Erkrankung hautgerichtete Therapien (Skin-directed Therapy; SDT) als First Line eingesetzt werden – und erst mit dem Fortschreiten der Erkrankung systemische (aggressivere) Behandlungsformen. Zu den wichtigsten SDT zählen Glukokortikoide, Chlormethin, Phototherapie (PUVA, Schmalband-UVB), lokalisierte Radiotherapie und Ganzhautbestrahlung (Total Skin Electron Beam; TSEB). Für alle hier angeführten Methoden sind gute bis sehr gute Ansprechraten in frühen Krankheitsstadien belegt – und auch die Möglichkeit einer Reinitiierung bei Rezidiven. Bei refraktärer und/oder fortgeschrittener Erkrankung kommen systemische Retinoide, Interferon-alpha, niedrigdosiertes Methotrexat, Chemotherapie, extrakorporale Photopherese, Alemtuzumab (Anti-CD52-Antikörper), Brentuximab-Vedotin (Anti-CD30-Antikörper-Toxin-Konjugat) und Mogamulizumab (Anti-CCR4-Antikörper) als Monotherapie oder in unterschiedlichen Kombinationen (mit SDT oder anderen Systemtherapien) zum Einsatz.
Die Therapie hängt auch von bereits verabreichten Vortherapien, der Präferenz und HRQoL der Patient:innen sowie von der Verfügbarkeit und Expertise ab und sollte, wenn möglich, an spezialisierten Zentren erfolgen. Das ultimative Therapieziel ist die Verbesserung und Erhaltung der Lebensqualität bei gleichzeitigem Streben nach Remission und Prävention von Krankheitsprogression.

Autorin:
Assoc. Prof.in PD Dr.in Constanze Jonak
Assoc. Prof.in PD Dr.in Constanze Jonak
Universitätsklinik für Dermatologie,
Medizinische Universität Wien

Ursprünglich erschienen:
AEK 11|2023
AEK 11|2023
Wissenswertes für die Praxis
- Differenzialdiagnosen der MF: mikrobielles oder atopisches Ekzem, Psoriasis vulgaris, Pityriasis rosea etc.
- Bei bis zu 30 % der MFPatient:innen kommt es zu extrakutanen Manifestationen.
- stadienhafter klinischer Verlauf der MF: von Patches über Plaques zu Tumoren sowie deren Koexistenz
Literatur:
Jonak C et al., J Dtsch Dermatol Ges 2021; 19(9): 1307–1335