






















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Bei der kardialen ATTR-Amyloidose kommt es in Folge von Transthyretin-Ablagerungen im Interstitium des Myokards zu einer Verdickung und Steifigkeit des Herzmuskels. Dank verbesserter Diagnosemöglichkeiten wird die seltene Erkrankung heute früher und auch häufiger erkannt. Der im Folgenden vorgestellte Fallbericht beschreibt typische Red Flags, die auf eine kardiale ATTR-Amyloidose hinweisen.
Klinik und Anamnese
Im Juli 2024 wurde ein 82-jähriger Patient zur weiteren Abklärung in die Amyloidose-Ambulanz des AKH Wien zugewiesen. Anlass waren eine progrediente Belastungsdyspnoe sowie eine erhöhte intraventrikuläre Septumdicke.
Klinisch konnte er entsprechend NYHA-Klasse II eingestuft werden. Der Verdacht auf eine Polyneuropathie bestand, konnte jedoch nicht bestätigt werden. Anamnestisch vermerkt wurde ein Zustand nach Karpaltunnelsyndrom-OP links vor 10 Jahren. Synkopen, eine Spinalkanalstenose sowie Beinödeme und AP-Symptomatik wurden negiert. Kardial bedingte Hospitalisierungen blieben bislang aus. Die Familienanamnese war hinsichtlich kardialer Erkrankungen unauffällig.
Diagnostik
Bei Erstvorstellung zeigte sich ein erhöhter NT-proBNP-Wert von 513 pg/ml, das hochsensitive Troponin T lag bei 38 ng/l und war damit ebenfalls leicht erhöht. Im EKG zeigte sich ein Sinusrhythmus und ein AV-Block 1.Grades. Die Blutdruckmessung ergab 130/74mmHg, die Herzfrequenz betrug 58BPM. Bezogen auf die Auskultation war der Patient unauffällig. Aufgrund der eingeschränkten Mobilität infolge eines Oberschenkelhalsbruches konnte der 6-Minuten-Gehtest zur Leistungsevaluierung nicht durchgeführt werden.
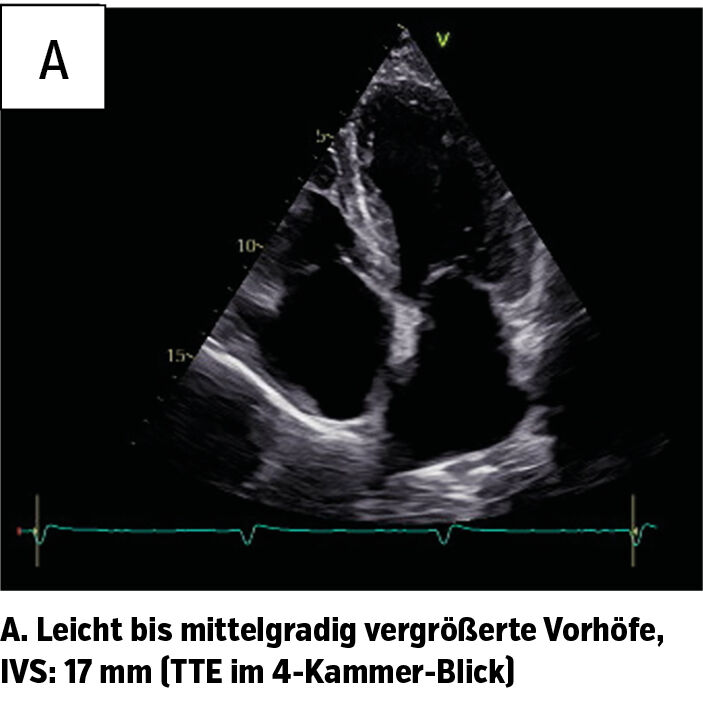
Im Rahmen der weiteren Diagnostik wurden eine MRT sowie eine TTE (transthorakale Echokardiografie) durchgeführt. In der TTE waren beide Vorhöfe leicht bis mittelgradig vergrößert (Abb. 1a), Aorten- und Mitralklappe zeigten jeweils eine leichte Verdickung. Ebenfalls konnte eine diastolische Ventrikelfunktionsstörung festgestellt werden. Eine Strain-Analyse wurde nicht durchgeführt. Typisch für eine Amyloidose ist hierbei ein Apical Sparing, auch als Cherry-on-Top-Phänomen bekannt.
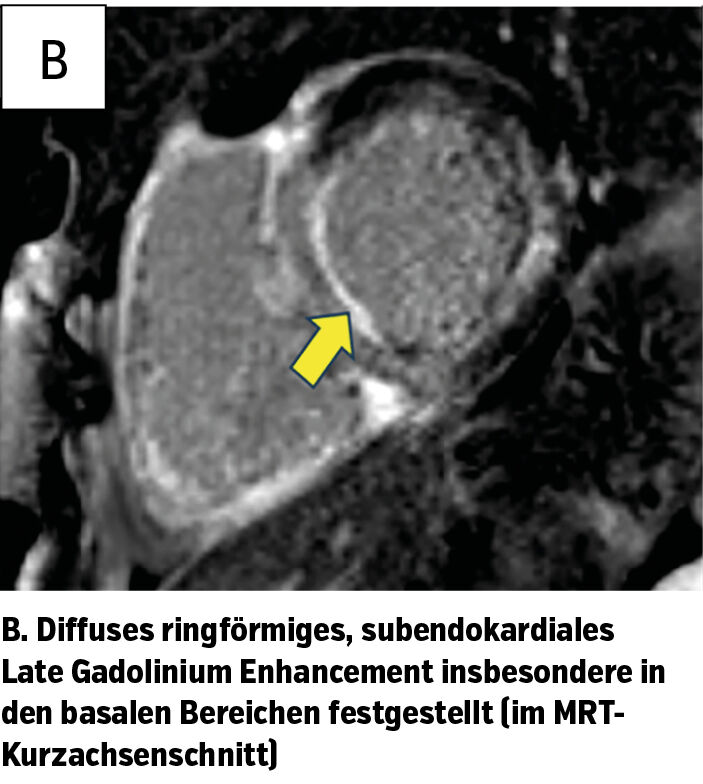
Insbesondere zeigte sich in der MRT das Vollbild einer kardialen Amyloidose (Abb. 1b). Ebenso auffällig war das erhöhte Extrazellulärvolumen, die septale Wanddicke und ein kleiner fokaler Perikarderguss. Diese Befundkonstellation lässt sich durch eine ausgeprägte diastolische Dysfunktion infolge der Amyloidablagerungen erklären – bei einer mäßiggradig eingeschränkten LVEF von 49%.
Weiters wurde eine Sequenzanalyse des TTR-Gens durchgeführt. Diese erbrachte keinen Nachweis einer pathogenen Mutation, sodass sich keine genetische Ursache für die Erkrankung feststellen ließ.
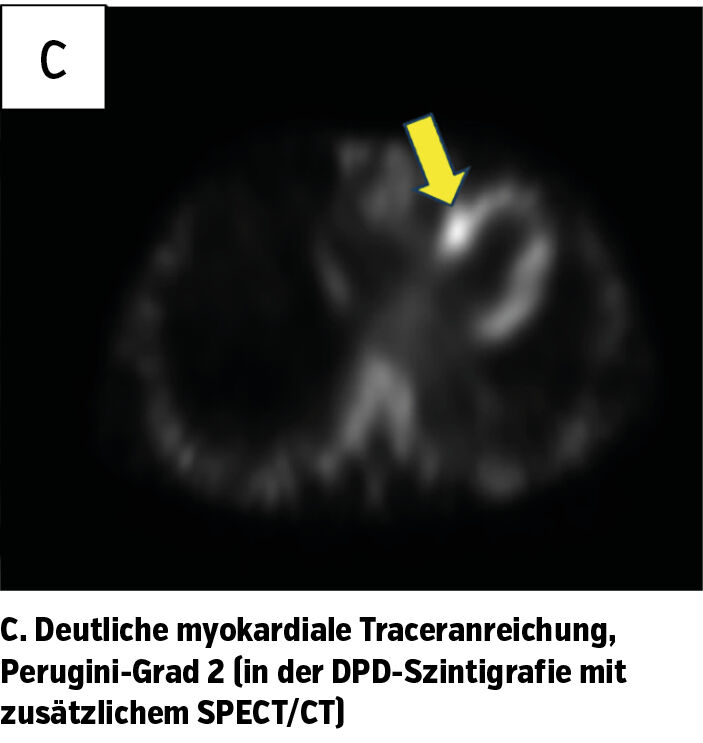
Zur weiteren Diagnosesicherung wurde eine DPD-Szintigrafie durchgeführt. Dabei zeigte sich eine deutlich erkennbare, myokardiale Traceranreicherung – entsprechend einem Perugini-Score von Grad 2 (Abb. 1c). Aufgrund dieses Ergebnisses, das auf eine Ablagerung von Amyloid im Myokard hindeutet, kann nach Ausschluss einer AL-Amyloidose die Diagnose einer Wildtyp-Transthyretin-Amyloidose gestellt werden.
Therapie
Bereits 2 Monate nach der Erstvorstellung wurde eine Behandlung mit Tafamidis eingeleitet. Tafamidis bewirkt eine Stabilisation von Transthyretin und verhindert eine Aufspaltung in ihre Untereinheiten.
Bei einer Verlaufskontrolle im Juni 2025 konnte der Zustand des Patienten als stabil beschreiben werden. Die Kurzatmigkeit konnte, bezogen auf die NYHA-Klassifikation, auf Grad I–II eingestuft werden, kardial bedingte Hospitalisierungen blieben weiterhin aus. Ein leichter Verlust der Muskelkraft und Kondition wurde vom Patienten beschrieben. Der 6-Minuten-Gehtest ergab eine Strecke von 307 m, was wiederum auf eine eingeschränkte Leistungsfähigkeit hinweist. Insgesamt konnte durch eine Tafamidis-Therapie eine Stabilisierung des Krankheitsbildes sowie eine leichte Verbesserung des Allgemeinzustandes erzielt werden.
Zusammenfassung
Zusammenfassend lässt sich sagen, dass dieser Fallbericht mehrere Red Flags aufzeigt. Das unterstreicht die Notwendigkeit, rechtzeitig „Rot zu sehen“. Anamnestische Indizien, wie beispielsweise ein Karpaltunnelsyndrom oder eine Spinalkanalstenose, treten häufig Jahre vor der Manifestation auf, werden allerdings nicht mit der Erkrankung Amyloidose in Verbindung gesetzt. Umso wichtiger ist es, dass bereits Orthopäd:innen bei diesen Problemen die Möglichkeit einer Amyloidose in Betracht ziehen und zumindest an die Durchführung einer Herzultraschalluntersuchung denken.
Weitere extrakardiale Hinweise sind Orthostasestörungen, gastrointestinale Beschwerden wie Diarrhö, Bizepssehnen-Rupturen, periorbitale Einblutungen sowie rasch fortschreitende Polyneuropathie. Kardial zeigt sich die Erkrankung charakteristischerweise durch Niedervoltagen im EKG. Auch häufig treten ein verlängertes QT-Intervall sowie Rhythmusstörungen wie Vorhofflimmern und AV-Blockierung auf.

Autorin:
Ap. Prof.in Priv.-Doz.in Dr.in Roza Badr Eslam
Ap. Prof.in Priv.-Doz.in Dr.in Roza Badr Eslam
Klinische Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

Autorin:
Cant. med. Stefanie Pahr
Cant. med. Stefanie Pahr
Klinische Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

Ursprünglich erschienen:
AEK 20|2025
AEK 20|2025
WAS PATIENT:INNEN WISSEN WOLLEN
- Wie ist die Prognose einer Amyloidose-Erkrankung mit vorwiegend kardialer Beteiligung?
Die medikamentöse Behandlung verbessert nachweislich sowohl die Prognose als auch die Lebenserwartung. Ein frühzeitiger Therapiebeginn führt zu einer Verringerung der Krankenhausaufnahmen sowie zu einer Verbesserung der Lebensqualität. - Gibt es weitere therapeutische Maßnahmen bei Verschlechterung des Allgemeinzustandes?
Oftmals ist es notwendig, die Herzinsuffizienztherapie zu optimieren und weitere Komorbiditäten zu behandeln.
Bildnachweis
Vorschaubild: © ChrWeiss – stock.adobe.com