





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Primäre Hyperoxalurie Typ 1
Seltene genetische Ursache für Nierensteine
20. März 2026
Ungefähr 10 % der Bevölkerung leiden unter Nierensteinen, mit zunehmender Prävalenz insbesondere in Industrieländern.1 Zu den metabolischen Risikofaktoren für Nierensteine zählen vor allem Adipositas und Diabetes.2 Bei einer kleinen Anzahl der Patient:innen mit Nephrolithiasis oder Nephrokalzinose ist jedoch eine monogenetische Variante die Ursache.
Fallbericht
Vorgeschichte und Anamnese. Ein 38-jähriger Mann soll an der Nephrologie Innsbruck wegen einer progredienten Verschlechterung seiner Nierenfunktion vorgestellt werden. Aufgrund eines Arbeitsunfalls mit multiplen Frakturen und Operationen nahm der Patient lange Zeit in höheren Dosen NSAR ein. Des Weiteren hatte er Oxalat-Nierensteine. Die Familienanamnese war negativ für Dialyse, Nierentransplantation und Nierensteine. Also lauteten unsere Differenzialdiagnosen: Analgetika-Nephropathie, CKD-assoziiert mit einer Nierensteinkrankheit, oder eine Oxalat-Nephropathie. Der Patient kam nicht zum Kontrolltermin, wurde aber notfallmäßig in seinem Heimatkrankenhaus mit einem akuten Nierenversagen und Anurie aufgenommen. Nach Beginn der Hämofiltration erfolgte umgehend eine Nierenbiopsie, und der Patient wurde nach Innsbruck überstellt.
Nierenbiopsie und Diagnose. Die Nierenbiopsie ergab die Diagnose einer renalen Oxalose mit Akkumulation von doppelbrechenden Kalziumoxalat-Kristallen in den Tubuli. Des Weiteren zeigten sich eine weit fortgeschrittene interstitielle Fibrose und Tubulusatrophie sowie eine interstitielle Nephritis, passend zur höchstwahrscheinlich terminalen Nierenerkrankung des Patienten (Abb.).
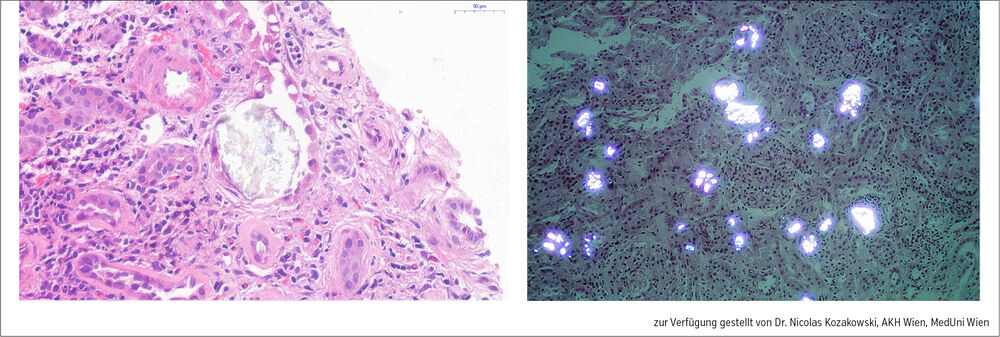
Abb.: Nierenbiopsie bei primärer Hyperoxalurie: linkes Bild: PAS-Färbung mit Kristallen im Tubulus; rechtes Bild: doppelbrechende Oxalat-Kristalle im polarisierten Licht
In der genetischen Analyse fand sich eine homozygote pathogene Mutation des AGXT-Gens, somit war die Diagnose gesichert: primäre Hyperoxalurie Typ 1 (PH1, siehe Kasten). In den weiteren Wochen wurden verschiedene Organbeteiligungen beim Patienten zusätzlich gesichert: Es zeigte sich eine kardiale Beteiligung, nachgewiesen mit kardialem MRT und einer Endomyokardbiopsie (Myokardbiopsat mit subendokardial akzentuierter interstitieller Oxalose), ein klinisch hochgradiger Verdacht auf eine Oxalat-Arthritis und eine sensomotorische, axonale Polyneuropathie.
Therapie. Unser Patient hatte eine homozygote AGT-Variante, die nicht responsibel auf Pyridoxin war. Einer Steigerung der Hämodialysefrequenz auf 6-mal/Woche wollte der Patient nicht zustimmen, immerhin konnten wir diese auf 4-mal 4 h/Woche erhöhen. Die stark erhöhten Plasmaoxalat-Werte (120–160µmol/l) sanken durch die Hämodialyse auf ca. 40 µmol/l ab (–67 bis –75 %), kehrten aber vor der nächsten Hämodialyse wieder auf den Ausgangswert zurück. Wir stellten die Indikation für eine Therapie mit Lumasiran und listeten den Patienten zur kombinierten Leber- und Nierentransplantation (LNTX). Das Ziel der Lumasiran-Therapie war eine Senkung der systemischen Oxalat-Last vor der geplanten Transplantation, um das Risiko einer Oxalat-Nephropathie im Nierentransplantat so niedrig wie möglich zu halten. Ebenso sollten die kardiale Oxalat-Belastung sowie die (vermutliche) Oxalat-Arthritis behandelt werden. Durch die Therapie mit Lumasiran konnte der Oxalat-Rebound um ca. 40–50 % reduziert werden, nach der Dialyse konnten Oxalat-Werte im Normalbereich (< 30 µmol/l) dokumentiert werden.
Als der Patient schließlich ein Organangebot bekam, erfolgten Hämodialysen unmittelbar vor der LNTX und täglich danach. Bei primärer Nichtfunktion des Nierentransplantates erfolgte eine Biopsie, die jedoch einen akuten Tubulusschaden mit unspezifischer Histomorphologie ohne Hinweise auf Abstoßung oder auf ein Rezidiv der Oxalose zeigte. In weiterer Folge erholte sich die Transplantatfunktion, das Serum-Kreatinin ca. 10 Monate nach der LNTX beträgt 1,0 mg/dl, der klinische Verlauf ist erfreulich unauffällig. Wir kontrollierten in unregelmäßigen Abständen die Harnoxalat-Ausscheidung, die sukzessive abnahm und sich nach ca. 6 Monaten im Normalbereich bewegte.
Fazit
PH1 ist eine sehr seltene autosomal-rezessive Erkrankung, die sich vor allem im Kindesalter mit Nierensteinen manifestieren kann. Eine LNTX und/oder eine Therapie mit Lumasiran (evtl. mit einer NTX) stellen therapeutische Möglichkeiten dar, die man mit den Betroffenen und der zuständigen Sozialversicherung diskutieren sollte.

Autor:
Priv.-Doz. Dr. Michael Rudnicki, FASN
Priv.-Doz. Dr. Michael Rudnicki, FASN
Universitätsklinik für Innere Medizin IV – Nephrologie und Hypertensiologie, Medizinische Universität Innsbruck

Ursprünglich erschienen:
AEK 05-06|2026
AEK 05-06|2026
PRIMÄRE HYPEROXALURIE TYP 1
Bei der PH1 ist das Enzym Alanin-Glyoxylat-Aminotransferase (AGT) defekt, das den Transfer von Aminogruppen zwischen Aminosäuren katalysiert. Durch den Defekt fällt vermehrt Glyoxylat an und wird durch die Laktatdehydrogenase zu Oxalat abgebaut. Solange die Nierenfunktion nicht oder nur wenig eingeschränkt ist (CKD-Stadien 1–3), wird das Oxalat über den Harn ausgeschieden, es fallen lediglich Oxalat-Kristalle oder Oxalat-Steine auf. Ab einer eGFR von 30–45 ml/min/1,73 m2 kann die Niere die Oxalat-Menge nicht mehr effektiv ausscheiden, es kommt zum Anstieg des Serumoxalats und zur Ablagerung von Oxalat hauptsächlich im Knochen, im Herzen und in den Gelenken.5
Obwohl die PH1 hauptsächlich im Kindesalter diagnostiziert wird, gibt es auch in 28 % der Fälle die erstmalige Diagnose im Erwachsenenalter. Leider findet sich bei 43 % der Fälle zum Zeitpunkt der Diagnose schon ein terminales Nierenversagen bzw. tritt eine Dialysepflicht im weiteren Verlauf bei 58 % der Patient:innen auf.6
THERAPIE DER PH1
Einerseits werden supportive Maßnahmen eingesetzt: Auf ausreichende Flüssigkeitsaufnahme ist zu achten (2–3 l/m² Körperoberfläche), und eine Steinprophylaxe mit Kaliumzitrat wird empfohlen. Andererseits wird Pyridoxin (Vitamin B6) als Kofaktor der AGT bei manchen AGT-Varianten mit einer Restenzymaktivität eingesetzt. An der Hämodialyse kann Oxalat zwar effektiv entfernt werden, allerdings kommt es zwischen den Dialysen zu einem Rebound, sodass eine intensivierte Dialyse empfohlen wird.
Eine LNTX stellte bis vor wenigen Jahren die State-of-the-Art-Behandlung dar. Dabei ist zu beachten, dass die neue Niere schlagartig beim Einsetzen der Nierenfunktion der ungeheuren Oxalat-Last des Körpers ausgesetzt ist, was akut zu einer Oxalat-Nephropathie führen kann. Deswegen müssen unmittelbar vor der Transplantation und in den Tagen danach tägliche Hämodialysesitzungen durchgeführt werden. Durch die neue Leber funktioniert der Oxalat-Metabolismus zwar wieder, jedoch kann die Oxalat-Ausscheidung noch 1–2 Jahre erhöht sein, weswegen erhöhte Trinkmenge und Kaliumzitrat weiterhin empfohlen werden.
WAHRSCHEINLICHKEIT FÜR SELTENE GENETISCHE URSACHEN BEI NIERENSTEINEN
In einer unselektierten Kohorte von 787 Nierensteinpatient:innen wurden umfassende genetische Analysen durchgeführt3: Bei 23 Patient:innen (3 %) fand sich eine monogenetische Mendel’sche Erkrankung (autosomal-rezessiv, autosomal-dominant oder X-chromosomal). Bei 18 der 23 Patient:innen war die Diagnose jedoch schon vor der Genetik bekannt, 5 Patient:innen wurden neu diagnostiziert: 3-mal Vitamin-D-24-Hydroxylase-Defizienz (CYP24A1), 1-mal primäre Hyperoxalurie Typ 3 (HOGA1) und 1-mal Bartter-Syndrom (SLC12A1). Insbesondere die 3 Patient:innen mit der homozygoten CYP24A1-Variante sind interessant, da bekannt ist, dass es bei diesen Patient:innen häufig polyzystische Nieren gibt, und tatsächlich lauteten die vorherigen Diagnosen autosomal-dominante polyzystische Nierenerkrankung.
Bei 64 Patient:innen (8 %) fanden sich heterozygote „Risikoallele“, d. h., es wurde eine pathogene oder wahrscheinlich pathogene Variante in nur einem Allel gefunden (bei einem autosomal-rezessiven Genotyp). Überwiegend handelte es sich um Varianten in Natrium-Phosphat-Kanälen SLC34A1, SLC34A3 und SLC9A3R1.
Insgesamt fand sich also bei 11 % aller Nierensteinpatient:innen ein pathologischer monogenetischer Befund. Der „diagnostic yield“ – also die Wahrscheinlichkeit, eine pathologische oder eine wahrscheinlich pathologische Variante zu finden, – liegt bei Kindern und jungen Erwachsenen zwischen 11 und 21 %, bei Erwachsenen sinkt dieser auf 1–11 %. Je jünger (< 25 Jahre), desto häufiger findet man eine genetische Erklärung für die Nierensteine.4 Unserer Erfahrung nach deuten auch häufige Rezidive auf das Vorliegen einer seltenen Ursache für Nierensteine hin.
Literatur:
Der Beitrag ist ursprünglich in
NephroScript 01/2025
erschienen.
NephroScript 01/2025
erschienen.
Bildnachweis
Vorschaubild: © Viktor - stock.adobe.com