


























































































Die österreichische Fachzeitschrift für Rheumatologie mit wissenschaftlichen Updates zu Pathogenese, Diagnostik und Therapie sowie DFP-Fortbildung in jeder Ausgabe.
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Die entzündlichen Darmerkrankungen (CED) Morbus Crohn (MC) und Colitis ulcerosa (CU) sind chronisch-remittierende, in ihrem Schweregrad progressive Erkrankungen des Verdauungstraktes, welche sich im Schub durch zum Teil hochfrequente Diarrhöen mit oder ohne Blutbeimengungen auszeichnen. Nach heutigem Wissensstand initiieren variable Umweltfaktoren auf Grundlage einer genetischen Suszeptibilität die Expression von klinisch unterschiedlichen Phänotypen, deren Endstrecke die Entzündung der Darmschleimhaut ist1. Im Falle der CU erstreckt sich die Entzündung anatomisch ausschließlich auf das Kolon, während MC den gesamten entodermalen Verdauungstrakt erfassen kann. Histologisch beschränkt sich der Entzündungsvorgang bei der CU auf die Tunica mucosa und submucosa, während bei MC die gesamte Darmwand betroffen ist.
Bislang lassen sich CED nur klinisch klassifizieren. Alter bei Erstdiagnose, Ausdehnung und Befallsmuster der Erkrankung (perianale Fistulierung bei Morbus Crohn) geben prognostische Hinweise. Im Wesentlichen versteht man die Ätiopathogenese von CED derzeit als Wechselspiel dreier Faktoren: Genetik, Mikrobiom und Umweltfaktoren.
Indirekte epidemiologische Hinweise haben schon lange einen genetischen Hintergrund für chronisch-entzündliche Darmerkrankungen vermuten lassen (Abb.). Nach ersten Zwillingsstudien in den 1980er-Jahren folgten in den 1990er-Jahren Linkage-Analysen und schließlich die Identifizierung erster Kandiatengene für IBD. Als Prototyp gilt die IL-10-Defizienz, welche zur Ausprägung eines schweren, fistulierenden Phänotyps von Morbus Crohn führt. Das Fehlen dieses immunregulatorischen Zytokins (oder dessen Rezeptors) ist jedoch bislang der einzige monogenetische Defekt, der eine chronisch-entzündliche Darmerkrankung bedingt2. Im Allgemeinen sind mehrere genetische Risikoallele von Nöten, um im Zusammenspiel mit Mikrobiom und Umgebungsfaktoren CED auszuprägen3.
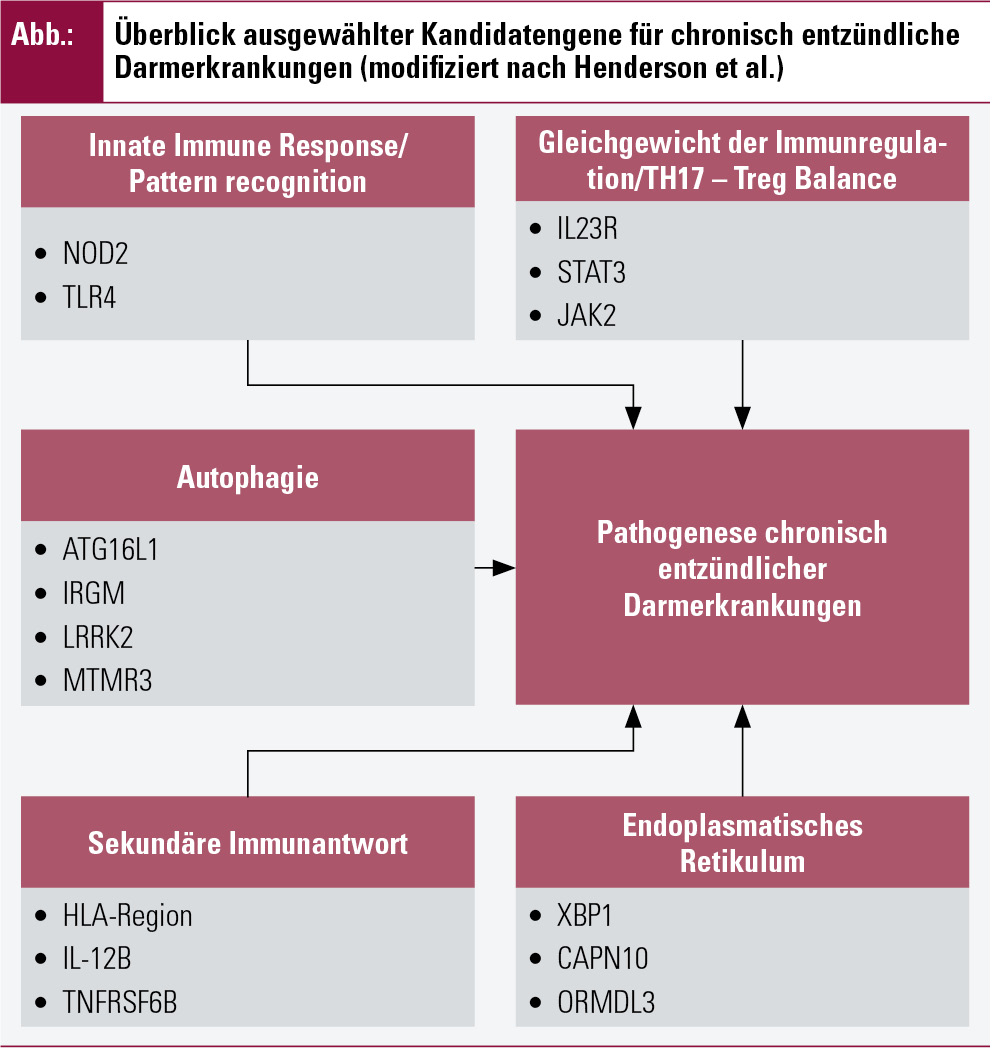
Risiko-Loci: Ein bedeutender Anteil der heute bekannten Suszeptibilitäts-Loci für Morbus Crohn und Colitis ulcerosa umfasst Gene, welche für den immunologischen Cross-Talk zwischen Mikrobiom und mukosalem Immunsystem verantwortlich sind. NOD2/CARD15 wurde bereits 1996 durch genetisches „fine mapping“ des IBD-1-Gens identifiziert und liefert bis heute das stärkste Signal in allen folgenden genomweiten Assoziationsstudien (GWAS)4. NOD2 kodiert für einen Pattern-Recognition-Rezeptor (PRR), welcher für die Erkennung von Muramyldipeptid, einem Bestandteil der Bakterienzellwand, benötigt wird5, 6, des Weiteren aber auch an der Aufrechterhaltung von Toleranzmechanismen und der Viruserkennung maßgeblich beteiligt ist6, 7.
Ein weiterer, für die Pathogenese der CED zentraler Zellvorgang ist der Prozess der Autophagie. Hierdurch werden nicht benötigte Zellorganellen, aber auch pathogene Mikroorganismen innerhalb des Zytoplasmas abgebaut. Dieser wird durch NOD2 via ATG16L1 induziert. Single-Nucleotide-Polymorphismen (SNP) konnten im Bereich von ATG16L1 nachgewiesen und mit CD assoziiert werden8.
Insgesamt konnten aus den kombinierten Daten der derzeit verfügbaren GWAS 163 Risiko-Loci für IBD, davon 110 übereinstimmende Risiko-Loci für CD und UC identifiziert werden9. Einzelne der rezent identifizierten Loci zeigen jedoch unterschiedliche Effekte bei CD und UC (PTPN22- und NOD2-Risiko für CD, jedoch protektiv bei UC). Trotzdem erklären alle bislang verfügbaren Daten nur 23 % der genetischen Varianz von CD und 16 % jener von UC10. Hochinteressant, wiewohl nicht ganz unerwartet, ist der signifikante gentische „overlap“, welcher IBD mit anderen immunmediierten Erkrankungen zeigt (z. B. Diabetes mellitus, ankylosierende Spondylitis, Psoriasis)9, welche zumindest zum Teil auch auf ähnliche pharmakologische Therapieansätze ansprechen.
Während Zwillingsstudien durchwegs als Argument für die Bedeutung einer genetischen Prädilektion herangezogen werden, weisen monozygote Konkordanzraten von 35 % bei CD und etwa 10 % bei CU in Wahrheit auf den überwiegenden Einfluss von Umgebungsfaktoren auf die Ausprägung des Phänotypes CED hin3. Rauchen, Ernährung, der Gebrauch oraler Kontrazeptiva, Appendektomie, Impfungen sowie virale und bakterielle Infektionen wurden in der Literatur beschrieben11.
Rauchen: Einzig das Zigarettenrauchen weist jedoch eine eindeutig reproduzierbare Effektgröße für die Genese von CED auf. Dieses korreliert bei Morbus Crohn mit höherer Krankheitsaktivität, dem Bedarf nach Immunsuppression und einer gesteigerten Wahrscheinlichkeit für intestinale Operationen, während Tabakrauch sich bei CU als protektiv erwiesen hat12. Weshalb dieser Einfluss besteht, ist letztlich unklar, mehrere mit Tabakrauchen assoziierte Veränderungen, vor allem des Mikrobioms wurden jedoch jüngst beschrieben13.
„Hygienehypothese“: Typisch für CED ist ferner ein deutliches Nord-Süd-Gefälle was Inzidenz- und Prävalenzraten betrifft. Die erhöhte und derzeit im Steigen begriffene Häufigkeit von CED im Norden ist auf Migranten übertragbar, welche nach Anpassung an den „westlichen Lebensstil“ der ansässigen Bevölkerung vergleichbare Häufigkeiten für CED aufweisen. Dieses Phänomen hat schon Ende der 1980er-Jahre zur Formulierung der so genannten „Hygienehypothese“ geführt, der zufolge eine zu geringe Exposition gegenüber Parasiten und anderen Infektionserregern durch verbesserte sanitäre Bedingungen zu einer Fehlentwicklung des Immunsystems und letztlich zu (Auto-)Immunerkrankungen, darunter auch CED, führt14, 15.
Ohne Bakterien keine CED. Dieser Grundsatz wurde bereits früh in der Erforschung der chronisch-entzündlichen Darmerkrankungen geprägt und hat nichts von seiner Gültigkeit verloren16. Tatsächlich entwickeln Versuchstiere, welche unter völlig keimfreien Bedingungen gehalten werden, keine der experimentellen Modellformen der Colitis17.
Die Prädilektionsstellen für Morbus Crohn im Bereich des terminalen Ileums und des Kolons sind jeweils Hotspots der Auseinandersetzung des mukosalen Immunsystems und der Mikroflora10, wodurch zumindest bei bestimmten Subgruppen von Patienten durch die Reduktion der bakteriellen Dichte durch den Einsatz von Breitspektrumantibiotika (z. B.: Ciprofloxacin) eine klinische Besserung erreicht werden kann18. Endogene antibakterielle Peptide, die so genannten Defensine, werden bei Patienten mit ilealem Morbus Crohn in geringeren Konzentrationen sezerniert als bei Kontrollen19.
Dysbiose: Ferner wurde bei Patienten mit IBD im Vergleich zu gesunden Kontrollen eine ausgeprägte Reduktion der Biodiversität der Mikroflora im Allgemeinen und eine Verschiebung von den beim Gesunden dominanten Firmicutes zu den Proteobacteria im Speziellen beobachtet20. Hierdurch entsteht möglicherweise eine ökologische Nische für Pathogene, welche dann durch eine dauerhafte Besiedlung zur chronischen Entzündung der Schleimhaut führen. Tatsächlich haben mehrere Arbeitsgruppen sowohl mit klassischen Kultur- als auch mit molekularbiologischen Methoden eine Kolonisation durch pathogene, enteroinvasive E. coli bei Patienten mit IBD nachgewiesen21, 22.
Der Grund für diese markanten Verschiebungen ist bislang ungeklärt. Jedoch konnten inzwischen auch genetische Faktoren identifiziert werden, welche durch ihre Funktion mit dem Phänomen der Dysbiose in Verbindung gebracht werden können. Besondere Aufmerksamkeit genießt diesbezüglich der Prozess der Autophagie, durch welchen Zellorganellen, aber auch Mikroorganismen („Xenophagie“) intrazellulär abgebaut werden23. Durch die Expression von Risikovarianten oder veränderte intrazelluläre Konzentrationen der beteiligten Proteine (ATG16L1, IRGM, NOD2) ist dieser Abbau ineffektiv, was wohl im Konzert mit weiteren Stimuli letztlich zur mukosalen Entzündung führt10.
Insgesamt muss bei der Pathogenese von Morbus Crohn und Colitis ulcerosa von einem „Multi-Hit-Modell“ ausgegangen werden, bei dem auf der Grundlage eines oder mehrerer genetischer Risikomerkmale bestimmte Einflussfaktoren auf das Ökosystem Darm einwirken und dadurch den Phänotyp Morbus Crohn oder Colitis ulcerosa zur Ausprägung bringen. Die große Herausforderung der kommenden Jahre wird es sein, die Erkenntnisse aus Genetik, Epidemiologie und Mikrobiologie zu vereinheitlichen. Zukünftige Therapiekonzepte müssen wohl der großen Diversität an „CED Syndromen“ oder „Subtypen“ Rechnung tragen, welche sich unter den Oberbegriffen Morbus Crohn und Colitis ulcerosa einreihen lassen und ihre Ansatzpunkte an die jeweils dominante Dysregulation der Immunhomöostase des einzelnen Patienten anpassen.

Klinische Abteilung für Gastroenterologie und Hepatologie, Universitätsklinik für Innere Medizin III, Medizinische Universität Wien

Klinische Abteilung für Gastroenterologie und Hepatologie, Universitätsklinik für Innere Medizin III, Medizinische Universität Wien


