


























































































Die österreichische Fachzeitschrift für Rheumatologie mit wissenschaftlichen Updates zu Pathogenese, Diagnostik und Therapie sowie DFP-Fortbildung in jeder Ausgabe.
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Kennzeichen dieser sich meist im Kindesalter manifestierenden Erkrankungen sind neben den Fieberschüben, die allerdings manchmal auch völlig fehlen können, entzündliche Begleitsymptome an Haut, Schleimhaut, serösen Häuten und Gelenken1, 2. Diese sind serologisch von einer ausgeprägten Entzündungsreaktion mit Erhöhung von Serum-Amyloid A begleitet, was auf das zum Teil erhebliche sekundäre Amyloidoserisiko hinweist.
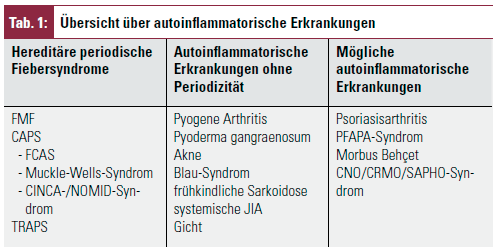
Seit 1997 die ersten genetischen Ursachen für zwei hereditäre Fiebersyndrome, das familiäre Mittelmeerfieber (FMF)3 und das sog. „Hibernian fever“4 entdeckt und dadurch diese Erkrankungsgruppe als eigene Entität wahrgenommen wurde, hat sich in der Begriffsbezeichnung der früher synonym verwendeten Begriffe die Einteilung geändert. Heute stellen die hereditären Fiebersyndrome eine Untergruppe der autoinflammatorischen Erkrankungen dar (Tab. 1).
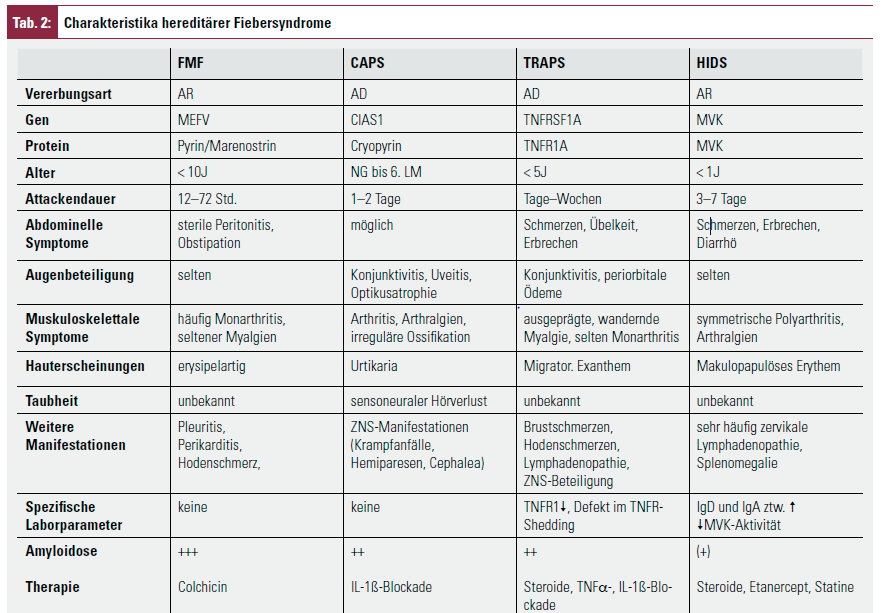
Hereditäre Fiebersyndrome sind monogenetische Erkrankungen, die durch Mutationen von Genen verursacht sind, die für die Kodierung von Proteinen zuständig sind, die bei der Regulation von entzündlichen Prozessen eine Schlüsselrolle spielen. Zu ihnen werden heute das Familiäre Mittelmeerfieber (FMF), das TNF-Rezeptor-1-assoziierte periodische Syndrom (TRAPS), das cryopurinassoziierte periodischen Syndrom (CAPS) sowie das Hyper-IgD-Syndrom (HIDS) gezählt (Tab. 2).
Der Begriff „Inflammasom“ beschreibt zytoplasmatische Komplexe aus multiplen Proteinen, die die Aktivierung von inflammatorischen Caspasen vermitteln. Caspasen sind Proteasen, die in apoptotische oder inflammatorische Signalwege involviert sind. Das Inflammasom kann durch infektiöse Trigger, aber auch endogen (z. B. durch Harnsäurekristalle, wie bei der Gicht) aktiviert werden. Bei den hereditären Fiebersyndromen kommt es durch Genmutationen zu einer Autoaktivierung des Inflammasoms und damit zu einer Überproduktion von IL-1β.
Die vorwiegend autosomal rezessiv vererbte Erkrankung ist durch Mutationen im MEFV-Gen3 verursacht. Die Erkrankung hat eine typische ethnische Verteilung und wird bei Patienten aus dem östlichen Mittelmeerraum besonders häufig (Prävalenz in der Türkei 1 : 500–1000), aber auch bei uns zunehmend gefunden5. Die selbstlimitierenden febrilen Attacken dauern 12–72 Stunden und sind von Zeichen der Serositis (Peritonitis, Pleuritis und Arthritis) begleitet6. Myalgien sind häufig. Erysipelartige Hautveränderungen, Entzündungen der Tunica vaginalis testis, sterile Meningitiden und protrahiert verlaufende Arthritiden sind selten. Das Amyloidoserisiko für unbehandelten Patienten beträgt 60–80 %6, 7. Bei adäquater prophylaktischer Therapie mit Colchizin (0,5 mg/d bei Patienten < 5 a, 1 mg/d bei Patienten von 5–10 a, 1,5 mg/d ab 10 a, einschleichend bis zu einem Tagesmaximum von 2 mg) sind 2/3 der Patienten schubfrei8, die übrigen bis auf etwa 2–3 % deutlich gebessert. Bei diesen Nonrespondern kann eine gegen IL-1β gerichtete Therapie versucht werden.
Unter dem Begriff CAPS werden die früher als separate Entitäten geführten Erkrankungen familiäre Kälteurtikaria (FCU), das Muckle-Wells-Syndrom (MWS) und das Chronic infantile neurological cutaneous and articular syndrome/Neonatal-onset multisystem inflammatory disease (CINCA-/NOMID-Syndrom) subsumiert, seit bekannt ist, dass alle drei auf einer Mutation im CIAS1-Gen beruhen9. Sie sind sehr selten (1–2 Fälle/Mio./a) und manifestieren sich neonatal bzw. in den ersten Lebensmonaten. Sie werden heute als unterschiedlich schwere Krankheitsverläufe ein und derselben Erkrankung angesehen, wobei der Schweregrad von der FCU über das MWS zum CINCA-/NOMID-Syndrom hin zunimmt und auch das Amyloidoserisiko in derselben Reihenfolge steigt.
Die gemeinsamen klinischen Charakteristika sind hohes Fieber, urtikarieller Ausschlag, Konjunktivis, Hörminderung und
Kennzeichen dieser sich meist im Kindesalter manifestierenden Erkrankungen sind neben den Fieberschüben, die allerdings manchmal auch völlig fehlen können, entzündliche Begleitsymptome an Haut, Schleimhaut, serösen Häuten und Gelenken1, 2. Diese sind serologisch von einer ausgeprägten Entzündungsreaktion mit Erhöhung von Serum-Amyloid A begleitet, was auf das zum Teil erhebliche sekundäre Amyloidoserisiko hinweist.
Seit 1997 die ersten genetischen Ursachen für zwei hereditäre Fiebersyndrome, das familiäre Mittelmeerfieber (FMF)3 und das sog. „Hibernian fever“4 entdeckt und dadurch diese Erkrankungsgruppe als eigene Entität wahrgenommen wurde, hat sich in der Begriffsbezeichnung der früher synonym verwendeten Begriffe die Einteilung geändert. Heute stellen die hereditären Fiebersyndrome eine Untergruppe der autoinflammatorischen Erkrankungen dar (Tab. 1).
Hereditäre Fiebersyndrome sind monogenetische Erkrankungen, die durch Mutationen von Genen verursacht sind, die für die Kodierung von Proteinen zuständig sind, die bei der Regulation von entzündlichen Prozessen eine Schlüsselrolle spielen. Zu ihnen werden heute das Familiäre Mittelmeerfieber (FMF), das TNF-Rezeptor-1-assoziierte periodische Syndrom (TRAPS), das cryopurinassoziierte periodischen Syndrom (CAPS) sowie das Hyper-IgD-Syndrom (HIDS) gezählt (Tab. 2).
Der Begriff „Inflammasom“ beschreibt zytoplasmatische Komplexe aus multiplen Proteinen, die die Aktivierung von inflammatorischen Caspasen vermitteln. Caspasen sind Proteasen, die in apoptotische oder inflammatorische Signalwege involviert sind. Das Inflammasom kann durch infektiöse Trigger, aber auch endogen (z. B. durch Harnsäurekristalle, wie bei der Gicht) aktiviert werden. Bei den hereditären Fiebersyndromen kommt es durch Genmutationen zu einer Autoaktivierung des Inflammasoms und damit zu einer Überproduktion von IL-1β.
Die vorwiegend autosomal rezessiv vererbte Erkrankung ist durch Mutationen im MEFV-Gen3 verursacht. Die Erkrankung hat eine typische ethnische Verteilung und wird bei Patienten aus dem östlichen Mittelmeerraum besonders häufig (Prävalenz in der Türkei 1 : 500–1000), aber auch bei uns zunehmend gefunden5. Die selbstlimitierenden febrilen Attacken dauern 12–72 Stunden und sind von Zeichen der Serositis (Peritonitis, Pleuritis und Arthritis) begleitet6. Myalgien sind häufig. Erysipelartige Hautveränderungen, Entzündungen der Tunica vaginalis testis, sterile Meningitiden und protrahiert verlaufende Arthritiden sind selten. Das Amyloidoserisiko für unbehandelten Patienten beträgt 60–80 %6, 7. Bei adäquater prophylaktischer Therapie mit Colchizin (0,5 mg/d bei Patienten < 5 a, 1 mg/d bei Patienten von 5–10 a, 1,5 mg/d ab 10 a, einschleichend bis zu einem Tagesmaximum von 2 mg) sind 2/3 der Patienten schubfrei8, die übrigen bis auf etwa 2–3 % deutlich gebessert. Bei diesen Nonrespondern kann eine gegen IL-1β gerichtete Therapie versucht werden.
Unter dem Begriff CAPS werden die früher als separate Entitäten geführten Erkrankungen familiäre Kälteurtikaria (FCU), das Muckle-Wells-Syndrom (MWS) und das Chronic infantile neurological cutaneous and articular syndrome/Neonatal-onset multisystem inflammatory disease (CINCA-/NOMID-Syndrom) subsumiert, seit bekannt ist, dass alle drei auf einer Mutation im CIAS1-Gen beruhen9. Sie sind sehr selten (1–2 Fälle/Mio./a) und manifestieren sich neonatal bzw. in den ersten Lebensmonaten. Sie werden heute als unterschiedlich schwere Krankheitsverläufe ein und derselben Erkrankung angesehen, wobei der Schweregrad von der FCU über das MWS zum CINCA-/NOMID-Syndrom hin zunimmt und auch das Amyloidoserisiko in derselben Reihenfolge steigt.
Die gemeinsamen klinischen Charakteristika sind hohes Fieber, urtikarieller Ausschlag, Konjunktivitis, Hörminderung und Arthropathien.
Beim CINCA-/NOMID-Syndrom bestehen häufig zusätzliche charakteristische morphologische Auffälligkeiten, wie eine vorgewölbte hohe Stirn und eine Sattelnase. Symptome der ZNS-Inflammation reichen von Kopfschmerzen bis zu Krampfanfällen und psychomentaler Retardierung. Eine sensorineurale Taubheit infolge der Cochleainflammation und eine Neuritis nervi optici mit Papillenödem und Erblindungsgefahr bestehen im späteren Lebensalter. Unbehandelt beträgt das Mortalitätsrisiko im Kindesalter 20 %.
Da IL-1 die zentrale Rolle in der Pathogenese von CAPS spielt, sind Substanzen, die IL-1 blockieren, wirksam. Zugelassen zur Behandlung von CAPS ist Canakinumab, ein humaner monoklonaler IgG1-Anti-IL-1β-Antikörper10. Empfohlen sind 2 mg/kg bei Patienten unter 40 kg einmalig alle 8 Wochen s. c. oder max. 150 mg/Injektion.
Der früher als familiäres Hibernienfieber bezeichneten Erkrankung liegt eine autosomal dominante Mutation des TNFRSF1A- Gens zugrunde, die für den extrazellulären Teil des TNFRp55 kodiert. TNFR1 ist ein membrangebundener Rezeptor, der auf den meisten Zellen synthetisiert wird. Durch sog. „shedding“ werden lösliche Rezeptoren gebildet, die TNF-α inaktivieren und dadurch den Grad der Entzündung durch kompetitive Rezeptorblockade reduzieren. TRAPS beginnt in der frühen Kindheit mit Fieberschüben über 1–4 Wochen ohne Periodizität. Häufige Begleitsymptome sind abdominelle und thorakale Schmerzen durch Serositis. Charakteristisch sind ausgeprägte wandernde Myalgien durch eine begleitende Fasziitis. Gelenksbeteiligungen kommen als Arthralgien und Arthritiden vor. Augenbeteiligung mit Konjunktivitis und periorbitalen Ödemen sind ebenfalls typisch. Weitere Hautveränderungen sind ausgeprägte, überwärmte Erytheme an Stamm und Extremitäten.
Das Amyloidoserisiko liegt bei 25 %. Etanercept (0,8 mg/kg/Woche s. c.)11 wirkt in den meisten Fällen. In einer Pilotstudie zeigte sich auch die Wirksamkeit von Anakinra12.
Ursache der autosomal rezessiv vererbten Erkrankung sind Mutationen des MVK-Gens, das für das Enzym Mevalonatkinase kodiert und zu einer verminderten Enzymaktivität führt13. Die genauen Mechanismen, die zu den autoinflammatorischen Symptomen führen, sind unbekannt. Charakteristisch sind alle 4-8 Wochen auftretende Fieberattacken, die nach einigen Tagen wieder abklingen. Trigger können Impfungen, virale Infekte oder Stress sein. Begleitsymptome sind abdominelle Beschwerden, wie Erbrechen und Diarrhö, eine Lymphadenopathie, Splenomegalie, Arthralgien und Arthritiden sowie ein makulopapulöses oder urtikarielles Exanthem.
Serologisch wegweisend sind die zumeist erhöhten IgD- und IgA-Spiegel und eine verminderte Mevalonatkinaseaktivität. Amyloidose kommt sehr selten vor.
Therapeutisch kommt eine einmalige Prednison-Gabe (1 mg/ kg) zum Einsatz. IL1-Antagonisten, Anti-TNF-α-Therapie und Simvastatin wurden erfolgreich angewandt.
Patienten mit hereditären Fiebersyndromen präsentieren sich klinisch sehr vielfältig und stellen dadurch eine große diagnostische Herausforderung für Mediziner unterschiedlichster Fachgebiete dar. Durch Aufklärung der zugrunde liegenden Pathomechanismen und Verfügbarkeit von spezifisch wirksamen Medikamenten konnte in den letzten Jahren eine enorme Lebensqualitätssteigerung für diese Patienten erzielt werden.
1) Drenth JPH, van der Meer JWM, Hereditary periodic fever. N Engl J Med 2001; 345:1748–1757.
2) Stojanov S, Kastner DL, Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol 2005; 17:586–599.
3) The international FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial mediterranean fever. Cell 1997; 90(4):797–807.
4) McDermott MF, Aksentijevic et al., Germline mutations in the extracellular domains of the 55kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97(1):133–44.
5) Ben-Chetrit E, Levy M, Familial Mediterranean fever. Lancet 1998; 351(9103):659–64. 6) Sohar E, Gafni J et al., Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43(2):227–53.
7) Özdemir AL, Sökmen C, Familial Mediterranean fever among the Turkish people. Am J Gastroenterol 1996; 51 (4):311–6.
8) Kallinich T, Haffner D, Niehues T et al., Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics 2007; 119(2):e474–83.
9) Feldmann J, Prieur AM, Quartier P et al., Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 71(1):98–203.
10) Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB et al., Use of Canakinumab in the Cryopyrin-Associated Periodic Syndrome. N Engl J Med 2009; 360(23);2416–2425. 11) Hull KM, Drewe E, Aksentijevich I et al., The TNF-receptor associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81(5):349–368.
12) Gattorno M, Pelagatti MA, Meini A et al., Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 2008; 58(5):1516–1520.
13) Houten SM, Kuis W et al., Mutations in MVK, encoding mevalonate kinase, cause Hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet 1999; 22(2):175–7.


