























































































Ursprünglich erschienen:
UIM 08|2021
UIM 08|2021
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur RegistrierungDie Inzidenz der MGUS steigt mit zunehmendem Alter und tritt in etwa bei 5 % der Bevölkerung über 70 Jahre auf, wobei Männer etwas häufiger betroffen sind als Frauen. Das Risiko ist höher, wenn bei Verwandten ersten Grades bereits eine MGUS vorliegt. Eine Vorbeugung für das Entstehen einer MGUS ist nicht bekannt.
Üblicherweise wird das monoklonale Paraprotein zufällig im Rahmen einer Routinediagnostik mit Serum-Elektrophorese festgestellt. Obwohl die monoklonale Paraproteinämie als Präkanzerose gilt, gibt es derzeit kein etabliertes Programm zur Früherkennung. Sobald ein monoklonales Paraprotein nachgewiesen wird, gilt es durch weiterführende Untersuchungen eine zugrundeliegende Erkrankung zu diagnostizieren bzw. bei deren Ausschluss eine MGUS zu verifizieren. Dazu sind folgende diagnostische Mittel erforderlich:
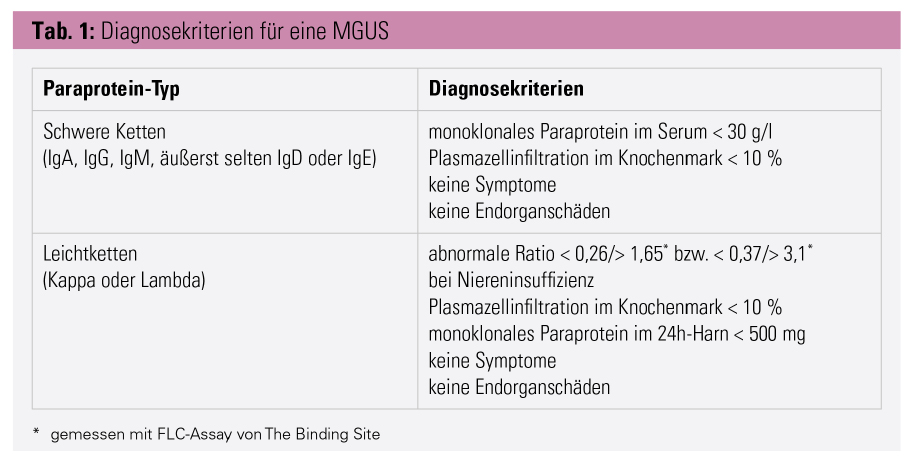
Ist die monoklonale Paraproteinämie im Serum nur diskret nachweisbar (IgG < 15 g/l bzw. IgA < 10 g/l bzw. Ratio der freien Leichtketten < 8), kann auf die Durchführung einer Knochenmarkbiopsie und bei Fehlen jeglicher skelettbezogener Symptome auf eine Bildgebung der Knochen verzichtet werden. Die International Myeloma Working Group (IMWG) hat für das Vorliegen einer MGUS die in Tabelle 1 angeführten Diagnosekriterien definiert.
Die MGUS kann in ein Multiples Myelom, in ein Lymphom, in einen Morbus Waldenström oder in eine AL-Amyloidose übergehen. Das Risiko für eine Progression hängt von verschiedenen Faktoren ab, die von Rajkumar et al. (Blood 2005; 106:812–17) wie folgt definiert wurden:
Wenn alle 3 Risikofaktoren vorliegen, besteht eine Hochrisiko-MGUS. Bei diesen Patienten liegt das Risiko für die Entwicklung einer Plasmazellneoplasie bei über 50 % nach 20 Jahren, während es bei MGUS-Patienten ohne Risikofaktoren nur bei etwa 5 % liegt.
Die Abgrenzung der MGUS von einer Plasmazellneoplasie ist essenziell und durch eine entsprechende Diagnostik möglich.
Beim Nachweis eines monoklonalen Paraproteins ist das Erheben von CRAB-Kriterien (Hyperkalzämie, Niereninsuffizienz, Anämie und Knochenläsionen) und Myeloma-defining Events entscheidend (Tab. 2).

Sobald eines der CRAB-Kriterien erfüllt ist, liegt ein Multiples Myelom vor (Tab. 3). Bei der Abgrenzung zu einem Smouldering Multiplen Myelom sind die Infiltrationsdichte im Knochenmark und das Ausmaß der Paraproteinämie im Serum bzw. im 24h-Harn entscheidend (Tab. 3). Beim solitären Plasmozytom liegt eine einzelne Raumforderung mit bioptischem Nachweis von monoklonalen Plasmazellen bei gleichzeitig fehlender Knochenmarkinfiltration vor. CRAB-Kriterien und Myeloma-defining Events dürfen ebenfalls nicht gegeben sein.

Im Rahmen eines Morbus Waldenström besteht neben einer IgM-Paraproteinämie üblicherweise auch eine Lymphadenopathie, eine Splenomegalie und mitunter auch ein Hyperviskositätssyndrom. Typischerweise findet sich bei über 90 % der Patienten eine MYD88-Mutation der Plasmazellen.
Eine AL-Amyloidose manifestiert sich mit unterschiedlichen Symptomen je nach betroffenen Organen. So kann es etwa zum Auftreten einer restriktiven Kardiomyopathie, Albuminurie, sensomotorischen Neuropathie, Splenomegalie, Makroglossie sowie zu Hauteinblutungen und Diarrhöen kommen. Laborchemisch sollen bei suspizierter AL-Amyloidose serologisch proBNP sowie die Albumin-Kreatinin- und Protein-Kreatinin-Ratio aus dem Spontanharn bestimmt werden. Ergänzend können eine Echokardiografie bzw. eine Magnetresonanztomografie des Herzens sowie eine Sonografie der Nieren und Nerven durchgeführt werden. Bei suspektem Befund soll in weiterer Folge eine Biopsie des betroffenen Organs erfolgen.
Liegt eine monoklonale Paraproteinämie mit Niereninsuffizienz vor, die nicht auf das Vorliegen eines Multiplen Myeloms oder einer AL-Amyloidose zurückzuführen ist, muss eine monoklonale Gammopathie mit renaler Signifikanz (MGRS) in Erwägung gezogen werden. Diese kann nur durch eine Nierenbiospie verifiziert werden.
Bei MGUS besteht keine Therapieindikation. Sollte jedoch bereits eine Plasmazellneoplasie bestehen oder eine Grunderkrankung vorliegen, die mit dem monoklonalen Paraprotein assoziiert ist, muss die jeweilige Erkrankung therapiert werden. Nach Erstdiagnose einer MGUS soll eine erstmalige serologische Verlaufskontrolle nach 6 Monaten durchgeführt werden, um gegebenenfalls eine steigende Dynamik des monoklonalen Paraproteins feststellen zu können. Bei stabilen Parametern sollen im weiteren Verlauf jährliche Kontrollen erfolgen. Bei Patienten mit Niedrigrisiko-MGUS und Patienten mit sehr hohem Lebensalter kann eine ausschließlich symptomorientierte Verlaufskontrolle in Erwägung gezogen werden.
