


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Das Lungengewebe als Zielstruktur von rheumatologischen Systemerkrankungen
16. Juli 2012
Wir wollen hier das Augenmerk auf Diagnostik und Therapie der ILD (Inters – titial Lung Disease) bei häufigeren Krankheitsbildern der Rheumatologie legen. Sys – temvaskulitiden, die ebenfalls mit einer Lungenbeteiligung einhergehen, werden in einem Folgeartikel beschrieben.
Lungenbeteiligung bei einzelnen Krankheitsbildern
Zu der Krankheitsgruppe der Kollagenosen gehören der systemische Lupus erythematodes (SLE), die systemische Sklerose (SSc), das Sjögren-Syndrom (SS), die Polymyositis/Dermatomyositis (PM/DM), die Mixed Connective Tissue Disease (MCTD) und andere Overlapsyndrome, die sich aus Symptomkombinationen verschiedener Kollagenosen, aber auch der chronischen Polyarthritis (RA) ergeben.
Zu den lungenassoziierten Komplikationen zählen die Lungenfibrose (PF) als parenchymatöse Erkrankung, die pulmonalarterielle Hypertonie (PAH), die Pleuritis und Thromboembolien. PAH und PF sind gemeinsam mit einer Mortalität von über 50 % gefürchtete lebensbedrohende Komplikationen bei PatientInnen mit Kollagenosen. Die frühzeitige Diagnose ist daher sehr wichtig und umfasst meist eine Bronchoskopie mit BAL, Serologie, transthorakalen Lungenultraschall und natürlich die High-Resolution-Computertomografie (HRCT) als nicht-invasiven Goldstandard für die ILD-Diagnostik.3 Eine Lungenbiopsie ist nur mehr in seltenen, speziellen Fällen notwendig. Die transthorakale Lungensonografie zur Beurteilung des Lungengewebes und der Pleura ist eine neue Möglichkeit, Patienten mit bekannten Kollagenosen für eine Lungenbeteiligung im Rahmen einer ILD zu screenen. Aktuelle wissenschaftliche Beiträge berichten über eine Lungenbeteiligung bei der rheumatoiden Arthritis in 23 % der Fälle.4 Aus eigenen Studien wissen wir, dass wir ebenfalls bei 18 % der respiratorisch asymptomatischen RA-PatientInnen bereits Lungenveränderungen haben und dass wir diese mit transthorakalem Lungenultraschall detektieren können.
Der systemische Lupus erythematodes (SLE) hat eine Prävalenz von 25/100.000 Einwohnern. Vor allem Frauen im Alter zwischen 20.–40. Lebensjahr sind betroffen (w:m = 9:1). Die Ätiologie der Erkrankung ist bis dato ungeklärt, eine virale Genese sowie gestörte Abräumreaktion und Apoptose werden diskutiert. Der SLE ist eine Systemerkrankung, die sich auch durch Hautveränderungen (Schmetterlingsery – them), Arthropathie, Polyserositis, Alopezie, Glomerulonephritis, Vaskulitis und Blutbildveränderungen sowie durch eine Beteiligung des Knochenmarks auszeichnet. Eine gefürchtete Komplikation des SLE ist der Neurolupus, der auch als Primärsymptom der Erkrankung vorausgehen kann und oft als Psychose fehlinterpretiert wird. Da 35 % der PatientInnen über Thoraxschmerzen klagen, finden sich diese PatientInnen primär in orthopädischen oder unfallchirurgischen Ambulanzen wieder. ARDS oder diffuse hämorrhagische Alveolitis treten glücklicherweise nur in 1–5 % der Fälle auf. Serologisch finden sich in 96 % an- tinukleäre Antikörper (ANA). Erst ein Titer von > 1:160 sollte als positiv interpretiert werden. Subsets der ANA wie z. B. dsDNAAntikörper lassen auf die entsprechende Organbeteiligung schließen. Fehlen diese Antikörper, ist eine Organbeteiligung aber nicht auszuschließen. Weitere Subsets wie z. B. Smith-Antikörper (Sm) sind in der Diagnosefindung hilfreich (> Tab.).
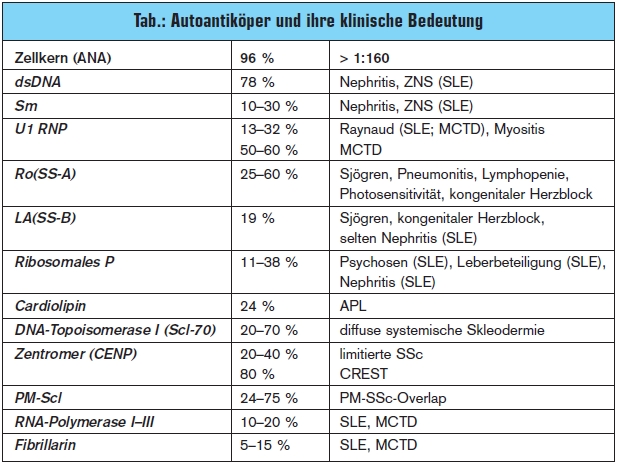
Die systemische Sklerose (SSc) zeichnet sich vor allem durch eine Vaskulopathie und Immunopathie aus. In den 1970er-Jahren war noch die renale Krise Haupttodesursache, heute sind es die progrediente Lungenfibrose und die PAH.5, 6 Durch den Einsatz von ACEHemmern und das Meiden von hohen Kortisondosen bei diesen PatientInnen ist die renale Krise in nur mehr ca. 1 % der PatientInnen zu beobachten.7 Eine Arbeitshypothese lässt darauf schließen, dass es zu einer überschießenden Aktivierung des Immunsystems durch einen Endothelschaden kommt. T-Lymphozyten sezernieren verschiedene Interleukine und TNF-alpha, wodurch es zu einer Aktivierung der Fibroblasten kommt, welche wiederum die Fibrosierung und Kollagenproduktion ankurbeln. B-Lymphozyten können Autoantikörper gegen z. B. Metalloproteinasen (MMP), welche eine überschießende Fibrosierung stoppen sollten, bilden. Die SSc betrifft vor allem Frauen zwischen dem 40. bis 50. und dem 60. bis 70. Lebensjahr. Die Ätiologie ist bis dato ungeklärt. Klinisch teilt man die SSc in die limitierte, die diffuse und die Sonderform, das CREST-Syndrom ein. Allen gemeinsam ist die Raynaud-Symptomatik und die Neigung zu digitalen Ulzerationen. Limitierte Formen haben ein höheres Risiko für die Entwicklung einer PAH und nur in seltenen Fällen einer Lungenfibrose. Das CREST-Syndrom (Calcinosis cutis, Raynaud-Syndrom, Ösophagusmotilitätsstörungen, Sklerodakytlie und Teleangiek – tasien) ist eine Sonderform. Diese Patienten haben ebenfalls ein höheres Risiko, eine PAH zu entwickeln. Serologisch finden sich in 10 bis 40 % der limitierten Formen und in bis zu 80 % der CREST-Syndrome Anti-Centromer- Antikörper (CENP). Die diffuse Form zeigt einen deutlich progressiveren Verlauf. Prinzipiell ist ein Befall aller inneren Organe möglich, die Lungenfibrose und der PAH sind aber die gefürchtetsten Komplikationen. Serologisch finden sich in bis zu 70 % Anti-Scl-70-Antikörper, die eine Diagnosefindung erleichtern.
Das Sjögren-Syndrom – auch Sicca-Syndrom – kann primär oder auch sekundär, d. h. vergesellschaftet mit anderen Kollagenosen, auftreten. Klinisch zeichnet sich diese Erkrankung durch Trockenheitsgefühl in den Augen („Sandkorngefühl“), Mund und Genitalbereich aus. Weiters klagen die PatientInnen über Obstipation und einen starken nächtlichen Trink – zwang. Eine seltene Komplikation ist die lymphozytäre Pneumonitis. Die Veränderungen können auch wie Pseudolymphome imponieren und sogar zu malignen Lymphomen transformieren. Serologisch finden sich in bis zu 80 % der Fälle Anti-Ro-Antikörper und Anti-La- Antikörper.
Die Mixed Connective Tissue Disease (MCTD, Sharp-Syndrom) ist eine undifferenzierte Kollagenose. Das klinische Bild ist sehr bunt (Myositis, Sklerodermie etc.), allen gemeinsam ist aber meist als Erstsymptom das Raynaud- Phänomen. Die MCTD mit Lungenbe – teiligung ist mit einer Mortalität von bis zu 40 % behaftet. Autoantikörper gegen U1RNP sind in ca. 60 % der Fälle zu finden.
Die Polymyositis und Dermatomyositis (PM/DM) ist eine autoimmunologische Entzündung der Muskulatur und ebenfalls sehr selten. Klinisch dominieren Muskelschwäche und Muskelatrophie sowie bei der DM die Besonderheit der lividroten Verfärbung der Oberlider. Der Muskelbefall ist meist symmetrisch und kann die unteren wie auch die oberen Extremitäten befallen. Das heißt aber nicht, dass ein asymmetrischer Befall eine Myositis ausschließt. Serologisch finden sich Antikörper gegen z. B. PM-Scl-70, sie sind aber generell sehr selten zu finden. Die Lunge kann ebenfalls im Sinne einer Fibrose befallen sein. In bis zu 40 % der PM und nur bis zu 10 % der DMPatienten finden wir Anti-Jo-1-(Synthetase)- Antikörper. Diese Antikörper bergen ein höheres Risiko für den Befall der Lunge.
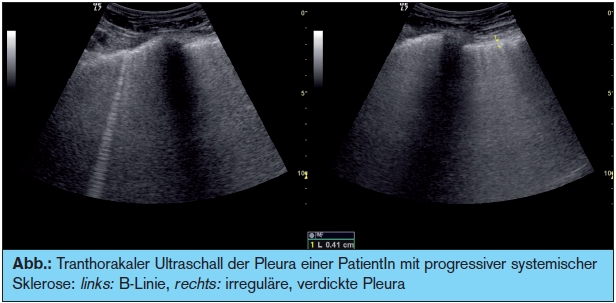
Bei chronischer Polyarthritis (RA) ist das Leitsymptom die Zerstörung der Gelenke durch einen die Knorpel und Knochen betreffenden erosiven Verlauf aufgrund der Proliferation der Synovia. Unbehandelt kann diese Erkrankung zur Invalidität führen. Die Freisetzung von Zytokinen, wie z. B. IL-1 oder TNF-alpha, führen zu einer Destruktion des Gewebes. Glücklicherweise kommen durch die frühzeitige Therapie mit Methotrexat, Leflunomid, Immunsuppressiva und den so genannten Biologika schwere Verlaufsformen nur mehr selten vor. Neue Diagnosekriterien und der frühzeitige Einsatz von Kombinationstherapien ermöglichen eine suffiziente Behandlung und ein besseres Outcome.8 Extraartikuläre Manifestationen der rheumatoiden Arthritis (RA) galten bis dato als eher selten. In aktueller Literatur wird berichtet, dass 23 % der PatientInnen einen Lungenbefall haben und dass besonders PatientInnen mit anticitrullinierten Antikörpern (aCCP) und positivem Rheumafaktor (IgA) eine ILD entwickeln können.4 Wir fanden sogar mittels Lungenultraschall, einer strahlenfreien Methode, bei 18 % respiratorisch beschwerdefreien RA-PatientInnen Lungenveränderungen, die sich mittels HRCT bestätigten (> Abb.). Interessant ist, dass unter TNF-Therapie granulomatöse Veränderungen in der Lunge bei RA-PatientInnen beschrieben wurden.9 Bis dato gibt es noch keine Studie, die zeigt, ob diese Veränderungen durch die Medikamente selbst induziert werden oder ob es bei bereits bestehender ILD zu einer Progredienz kommen kann. TNF-alpha- Blokker-Therapien werden bei idiopathischer ILD nicht eingesetzt, weil es zu einer Verschlechterung der Fibrose kommen kann.10–12
Diagnostik der Lungenmanifestationen
Diagnostisch ist das HRCT der nicht-invasive Goldstandard, um eine Alveolitis, dargestellt als milchglasartige Eintrübungen und bereits fibrotisch umgebautes Gewebe („Honeycombing“), darzustellen. Funktionelle Test wie die DLCO-Messung (Diffusing capacity of the Lung for Carbon monoxide) zur Detektierung restriktiver Störungen gehören zu den Basisuntersuchungen jeder diagnostischer Aufarbeitung. Besonders wichtig ist daher auch die Zusammenarbeit mit den Pulmonologen, um eine suffiziente Betreuung dieser Patienten zu gewährleisten. Die Diagnose einer Lungenbeteiligung ist nur in Zusammenhang mit der Grundkrankheit zu erstellen. Schwierig wird es, wenn sich keine weiteren Parameter finden und Patienten sich nur durch ein pathologisches HRCT und Dyspnoe präsentieren. In solchen Fällen kann eine Lungenbiopsie nützlich sein. Das bunte Bild und der uneinheitliche Verlauf dieser Rheumakrankheiten macht eine schnelle Diagnose oft unmöglich.
Therapie der Lungenmanifestationen
Therapeutisch stehen uns bei Lungenbeteili – gung im Rahmen von Autoimmunerkrankungen zur Induktionstherapie Glukokortikoide und Cyclophosphamid zur Verfügung. Zielführend ist eine medikamentöse Therapie vor allem im Stadium der frischen Entzündung. Ist die Fibroseentstehung einmal abgeschlossen, führen die Medikamente zu keiner wesentlichen Besserung der Klinik. Als Erhaltungstherapeutika werden vor allem Azathioprin, Mycophenolatmofetil oder Tacrolimus eingesetzt. Als Ultima Ratio bei therapieresistenten progredienten Lungenfibrosen bei Kollagenosen kann man die PatientInnen einer autologen Stammzelltransplantation unterziehen. Diese Maßnahme ist mit einer Mortalität von ca. 5 % behaftet. Vereinzelten Fallberichten zufolge führt Rituximab, ein Anti-CD20-Antikörper, zu einem Stagnieren der ILD bei SSc in cyclophospamidresistenten Fällen.13, 14 Wir setzen Rituximab erfolgreich bei Sklerodermie ein und beobachteten wiederholt eine Regredienz der Veränderungen im HRCT. Experimentell wirkt der Thrombininhibitor Dabigatran ebenfalls hemmend auf die Fibroblasten.
ZUSAMMENFASSEND wissen wir heute, dass die Haupttodesursache bei KollagenosepatientInnen die Lungenbeteiligung ist und dass die Früherkennung für das Ansprechen der Therapie und damit für das Outcome unserer PatientInnen entscheidend ist.
FACT-BOX
• Haupttodesursache ist bei 50 % der Kollagenosen die ILD und lungenassoziierte Komplikationen.
• 23 % der PatientInnen mit RA haben eine ILD.
• Frühzeitige Erkennung einer Lungenbeteiligung ist für das Outcome entscheidend.

Ursprünglich erschienen:
UIM 02|2012
UIM 02|2012
Herausgeber: Univ.-Prof. Dr. Günter J. Kreijs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2012-03-09
Zur Ausgabe »
Publikationsdatum: 2012-03-09
Zur Ausgabe »
