


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hämoglobinopathien – Die rasche Zunahme einer früher seltenen Erkrankung
16. August 2012
Hämoglobin (Hb), der rote Blutfarbstoff, ist das Protein mit der höchsten Konzentration im menschlichen Blut. Hb ist aus vier Proteinuntereinheiten, den Globinketten, aufgebaut, von denen jede in ihrem Inneren ein Häm, ein Porphyringerüst mit einem zentralen Eisenatom (Fe2+) (Eisen-Protoporphyrin IX), trägt, welches in Abhängigkeit von den äußeren Bedingungen Sauerstoff bindet oder abgibt und so den Sauerstofftransport bewerkstelligt. Die Bindungen zwischen den Globinketten sind nichtkovalent.
Humane Hämoglobine: Beim Mensch werden in der Embryonalzeit zunächst drei embryonale Hb-Typen gebildet, die mit Ende der Embryonalzeit durch drei andere Hb-Formen ersetzt werden. Diese sind aus α-, β-, γ- und δ-Globinketten aufgebaut; die Kettenlänge ist etwas unterschiedlich (α-Ketten 141, β-, γ- und δ-Ketten je 146 Aminosäuren), es besteht aber eine hohe Sequenzhomologie. Die Globinketten bilden die folgenden Hb-Moleküle: HbA bestehend aus zwei α- und zwei β-Ketten (α2β2)γγ, HbF bestehend aus zwei α- und zwei γ-Ketten (α2βγ2) und HbA2 bestehend aus zwei α- und zwei δ-Ketten (α2δ2) (Abb.). In den Erythrozyten findet sich in der Regel nicht eine Hb-Spezies, sondern ein Gemisch aus den drei Hb-Typen. Ihre Anteile am Gesamt-Hb unterscheiden sich allerdings deutlich: In der Fetalzeit überwiegt die γ-Ketten- und damit die HbF-Synthese. Als physiologische Ursache gilt der Umstand, dass die Sauerstoffaffinität von HbF größer ist als die von HbA und deshalb das fetale Blut jenen Sauerstoff aufnehmen und zum Gewebe transportieren kann, der vom HbA der Mutter bereits abgegeben wird und durch die Placenta diffundiert. Die Bildung von γ-Ketten und damit von HbF beim Fetus nimmt gegen Ende der Schwangerschaft zunächst langsam und dann um den Zeitpunkt der Geburt sowie postpartal rapide ab, während die Bildung von β-Ketten und damit von HbA gegenläufig deutlich ansteigt (Abb.). Nach etwa 12 bis 18 Monaten ist das endgültige Hämoglobinmuster gegeben, das zeitlebens weitgehend unverändert beibehalten wird: HbA: ca. 95–98 %, HbF: ca. 0,0–1,2 % und HbA2: ca. 1,5–3,3 %.
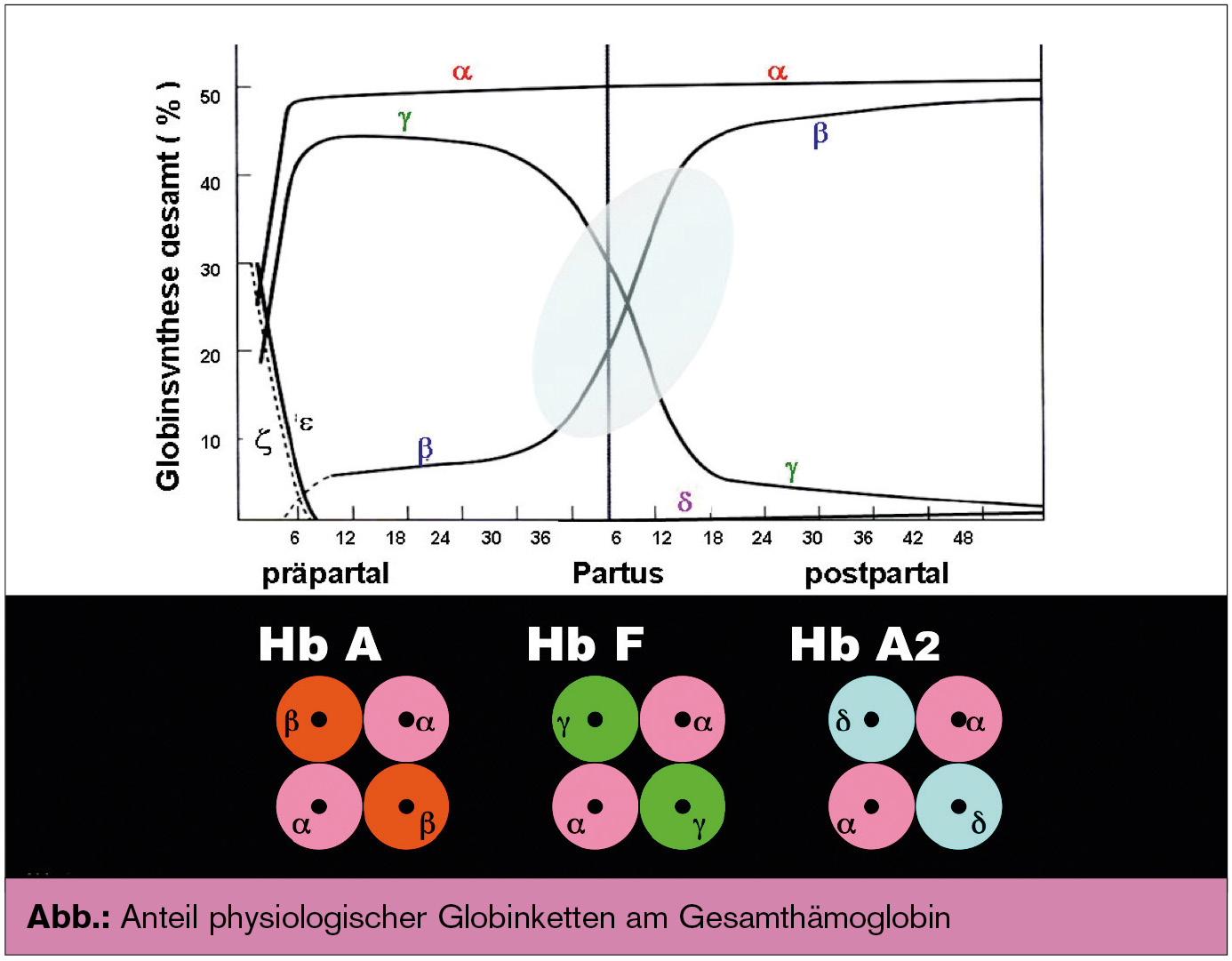
Genetik: Die Genloci der Globinketten befinden sich auf den Chromosomen 11 und 16. Am Chromosom 11 findet sich der sogenannte β-Globin-Gen-Cluster. Dieser enthält u. a. das β-Gen, das δ-Gen sowie zwei γ-Gene (G γ und Aγ); letztere kodieren γ-Ketten, die sich in einer Aminosäure unterscheiden (Glycin oder Alanin in Position 136); mit beiden γ-Kettenarten (Gγ und Aγ) kann HbF synthetisiert werden; der Anteil von Gγ- zu Aγ-Ketten im HbF des Erwachsenen beträgt etwa 2:3. Am Chromosom 16 sind jeweils zwei α-Gene (das α1- und α2-Gen) lokalisiert. Die beiden Gene sind auf eine Duplikation zurückzuführen und unterscheiden sich geringfügig in ihren Intronen, nicht aber in ihren Exonen; daher kodieren sie identische α-Globin-Ketten.
Hämoglobinopathien
Hämoglobinopathien sind angeborene Störungen der Bildung des Hämoglobins. Im Wesentlichen kann man zwei große Gruppen unterscheiden, Thalassämien und Hb-Varianten:
Thalassämien
Bei den Thalassämien besteht eine Störung der Bildung der physiologischen Globinketten. In Abhängigkeit vom zugrunde liegenden Defekt kommt es nur zu einer Verminderung („+“-Thalassämien) oder dem völligen Fehlen („0“-Thalassämien) des betroffenen Globinkettentyps. Dies bedingt einen relativen Überschuss der im normalen Ausmaß gebildeten Globinketten, denen komplementäre Globinketten zur Bildung normaler Hb-Moleküle fehlen. Diese überschüssigen freien Globinketten führen zur Hämolyse von Erythrozyten durch intraerythrozytäre Präzipitation oder Ablagerung an der Erythrozytenmembran. Damit verbunden kann eine massive Eisenfreisetzung mit konsekutiver Eisenüberladung und schweren Organschäden sein. Die Klinik von Thalassämien ist somit weniger auf die verminderte Hb-Bildung zurückzuführen, sondern auf die Erythrozytenschädigung verursacht durch die freien im normalen Ausmaß gebildeten Globinketten.
Die klinisch bei weitem wichtigsten Thalassämieformen sind jene, die die Synthese des HbA betreffen, also Störungen der Bildung der α-Globin-Ketten (α-Thalassämien) oder der β-Globin-Ketten (β-Thalassämien). Daneben gibt es weniger bedeutsame Thalassämieformen. Formal den Thalassämien zugerechnet werden meist auch jene Hämoglobinopathien, die durch die Synthese von Hybridglobinketten charakterisiert sind. Es sind das Globinketten, deren Aminosäuresequenz, bedingt durch ein pathologisches Crossingover, von zwei Globingenen kodiert wird. Die bekanntesten Vertreter sind die Lepore-Hämoglobine, die anstelle normaler β-Ketten über Globinketten verfügen, bei denen nur der C-terminale Teil vom β-Globin-Gen, der N-terminale Kettenabschnitt aber vom δ-Globin-Gen kodiert wird. Weiters werden vielfach die verschiedenen Formen der hereditären Persistenz von HbF den Thalassämien zugerechnet. Es sind das klinisch weitgehend asymptomatische Zustände mit einer lebenslang erhöhten HbF-Synthese.
Hb-Varianten
Bei diesen Hämoglobinopathien bestehen Mutationen der Globingene, die zur Synthese abnormaler Globinketten und in der Folge zur Bildung abnormaler Hämoglobinmoleküle führen. Besonders für die α- und die β-Ketten sind zahlreiche Mutationen beschrieben. Vorwiegend handelt es sich dabei um Punktmutationen, die zum Austausch einer Aminosäure führen (z. B. das Sichelzellhämoglobin [HbS], das HbC, HbD oder HbE). Wesentlich seltener sind Hämoglobinvarianten mit Globinketten abnormaler Kettenlänge sowie Globinketten, die mehrere mutierte Aminosäuren enthalten.
Genotyp und klinische Symptomatik: Als genetische Ursache von Hämoglobinopathien sind zurzeit rund 1.500 Mutationen beschrieben. Etwa 1.150 führen zur Synthese eines abnormalen Hb, die übrigen zu einer Thalassämie. Die Hämoglobinopathien können in heterozygoter Form (Anlage, „trait“) oder in homozygoter (bzw. doppelt heterozygoter) Form (Erkrankung, „disease“) vorliegen. So gut wie alle möglichen Kombinationsformen sind in der Realität beschrieben, z. B. das Zusammentreffen gleicher oder unterschiedlicher Thalassämieformen, die Kombination einer Thalassämie mit einer Hb-Variante oder die Kombination zweier verschiedener Hb-Varianten.
Die Symptomatik wird sowohl durch die Art der Hämoglobinopathie als auch durch den Genstatus bestimmt. So gibt es Formen, die im heterozygoten Zustand keine oder eine geringe Symptomatik aufweisen, und andere, die schon bei Heterozygotie zu schweren Krankheitsbildern führen. Umgekehrt gibt es Formen, die auch bei homozygoter Vererbung unauffällig verlaufen. Im Allgemeinen gilt, dass eine im heterozygoten Zustand bestehende Symptomatik bei homozygoter (oder doppelt heterozygoter) Vererbung oft deutlich verstärkt wird oder es erst dann zu klinisch relevanten Symptomen kommt. Ein gutes Beispiel ist das HbS: Heterozygote Personen sind meist asymptomatisch, bei homozygoten Patienten kann es infolge eines Absinkens des Sauerstoffpartialdrucks akut zu massiver Sichelung von Erythrozyten mit Durchblutungsstörung des Kapillarbettes und schwersten Schmerzzuständen sowie einer Reihe lebensbedrohlicher Komplikationen kommen (Sichelzellkrise). Es gibt aber auch Ausnahmen von der Regel: so werden die Symptome einer homozygoten β-Thalassämie durch Kombination mit einer α-Thalassämie abgemildert, weil bei dieser Kombination der durch die β-Thalassämie bedingte relative Überschuss an freien α-Globin-Ketten reduziert wird.
β-Thalassämien gehen großteils auf Punktmutationen zurück; im Gegensatz dazu sind die häufigsten α-Thalassämie-Formen durch Deletionen verursacht, die je nach Größe ein oder beide α-Gene eines Chromosoms 16 betreffen können. Personen, denen ein oder zwei α-Gene fehlen, sind meist beschwerdefrei. Beim Verlust dreier α-Gene spricht man von einer HbH-Erkrankung; ein solches Krankheitsbild kann auch resultieren, wenn 2 α-Gene wegen Punktmutationen ausfallen. Häufig, aber nicht zwingend, findet sich bei der HbH-Erkrankung eine erhöhte Erythrozytenzahl sowie zumeist eine Verminderung des Gesamthämoglobins, des MCV, MCH und des MCHC. Klinisch besteht meist eine mittelgradige Anämie mit deutlicher Mikrozytose und Hypochromasie. Nicht mit dem Leben vereinbar ist ein Fehlen aller vier α-Gene, da alle postpartalen Hämoglobine α-Ketten enthalten und somit keine normalen Hämoglobinformen gebildet werden können (Tab.). Die betroffenen Feten überleben meist bis ins 3. Trimenon mit Hilfe eines sonst nur embryonal gebildeten Hämoglobins (Hb Portland; ζ2γ2) undkommen als Frühgeburt zur Welt. Sie zeigen ein schweres klinisches Krankheitsbild, das als Hydrops fetalis Bart’s bezeichnet wird. Unbehandelt kommt es meist kurz nach der Geburt zum Exitus.
Epidemiologie: Hämoglobinopathien gehören global gesehen zu den häufigsten genetischen Störungen und betreffen weltweit hunderte Millionen Menschen, etwa 7 % der Weltbevölkerung. Sie sind besonders in Afrika, dem Mittelmeerraum und Südostasien weit verbreitet mit einer Frequenz, die in manchen Gebieten bei rund 30 % liegt. In Mittel- und Nordeuropa nimmt die Häufigkeit von Hämoglobinopathien infolge der modernen Bevölkerungsbewegungen schnell und stetig zu und liegt – mit regionalen Unterschieden – gegenwärtig bei etwa 1 % der Bevölkerung. Für Österreich gibt es keine systematischen Untersuchungen, doch dürfte der Anteil an Genträgern etwas niedriger, bei etwa 0,25 % der Bevölkerung, liegen. Allerdings ist ein weites Spektrum an Hämoglobinopathien vertreten. Trotz der großen Zahl der beschriebenen Hämoglobinopathieformen sind es nur einige wenige, die weltweit häufig vorkommen und global von epidemiologischer Bedeutung sind; ihr Hauptverbreitungsgebiet deckt sich mit den alten Malariagürteln der Erde, weil betroffene Personen gegen Malaria etwas resistenter sind und einen milderen Krankheitsverlauf zeigen. Zu nennen sind hier neben den α- und β-Thalassämien in erster Linie das HbS, das sich in verschiedenen Weltgegenden aufgrund von zumindest vier unabhängigen Mutationsereignissen verbreitet hat. Die genauen Resistenzmechanismen gegen Malaria sind allerdings bis heute nicht gesichert. Regional sehr häufig sind weiters das HbC, HbD und HbE.
Untersuchungsindikation und Klinik: In der Praxis ist in erster Linie bei ungeklärten mikrozytären Anämien, die auf Eisengabe nicht ansprechen (Beispiel: heterozygote Thalassämien), an eine Hämoglobinopathie zu denken. Bei seltenen Hämoglobinopathieformen kann allerdings eine ganz andere Symptomatik bestehen, z. B. eine schwere Hämolyse (instabile Hämoglobine), Polyglobulie oder Zyanose (Hämoglobine mit gestörter Sauerstoffaffinität). Eine weitere wichtige Untersuchungsindikation ist die Feststellung des Genstatus bei Paaren mit Kinderwunsch sowie die pränatale Diagnostik, falls beide Partner eine Hämoglobinopathie aufweisen, die bei kombinierter Vererbung zu einer schweren Erkrankung führen kann (Tab.). An eine Hämoglobinopathie sollte auch gedacht werden, wenn sich bei der HbA1c-Messung kein oder kein plausibler Messwert ergibt.

Therapie: Die Therapie der Hämoglobinopathien ist meist symptomatisch. Eine Heilung schwerer Formen ist gegenwärtig nur durch eine Knochenmark- oder Stammzelltransplantation möglich, gentherapeutische Ansätze haben bisher keinen Eingang in die klinische Routine gefunden. Bei polytransfundierten Patienten hat es im letzten Jahrzehnt große Fortschritte in der Eisenchelattherapie zur Verhinderung einer Eisenüberladung durch die Einführung oral wirksamer Präparate (Deferasirox, Deferipron) gegeben, die das lange bekannte, parenteral zu verabreichende Deferoxamin ergänzt oder abgelöst haben. Bei homozygoten β-Thalassämien und der HbS-Erkrankung wird teilweise erfolgreich versucht, den HbF-Anteil durch Reaktivierung der γ-Ketten-Produktion zu erhöhen; der Effekt beruht auf dem Umstand, dass bei β-Thalassämien HbF den Mangel an HbA zum Teil kompensieren kann und bei der HbS-Erkrankung, weil die γ-Ketten mit dem Prozess der Sichelzellbildung interferieren und so möglichen Sichelzellkrisen entgegenwirken. Das am längsten bekannte Präparat zur HbF-Erhöhung ist Hydroxyurea. Weiters sind Personen mit einer HbS-Erkrankung gegen Pneumokkoken und andere Erreger zu impfen und sollen zumindest im Kleinkindesalter eine kontinuierliche Antibiotikaprophylaxe erhalten.

Ursprünglich erschienen:
UIM 06|2012
UIM 06|2012
Herausgeber: Univ.-Prof. Dr. Günter J. Kreijs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2012-07-20
Zur Ausgabe »
Publikationsdatum: 2012-07-20
Zur Ausgabe »
