


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
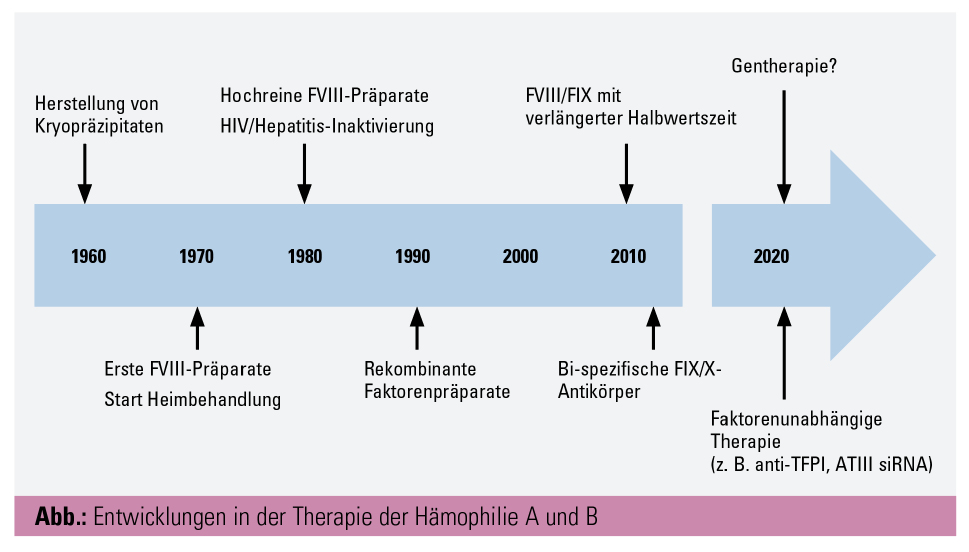
Entwicklungen in der Therapie der Hämophilie A und B
27. Mai 2019
Das generelle Ziel der Therapie der Hämophilie ist es, das – wegen des fehlenden Gerinnungsfaktors VIII (FVIII) oder IX (FIX) – bestehende Risiko schwerwiegender Blutungen zu reduzieren. Dies gelang in den letzten Jahren lediglich durch regelmäßige intravenöse Faktorensubstitution. Daten zu neuen Therapieansätzen geben jedoch Hoffnung, dass sich in naher Zukunft die Therapie der Hämophilie speziell im Hinblick auf die Belastungen für den Patienten signifikant verbessern wird.
Status quo: Hämophilie-Therapie in Österreich
Im österreichischen Hämophilie-Register schienen im Jahr 2016 753 Patienten und somit mehr als 85 % der zu erwartenden Anzahl von Hämophilen in Österreich auf – dabei verteilten sich 84,3 % der Patienten auf eine Hämophilie A und 15,7 % auf eine Hämophilie B (medianes Alter: 34 Jahre). 294 (39,0 %) der Patienten wiesen dabei eine schwere Form mit einem Faktor VIII/IX von <1 % auf. Von diesen Patienten mit schwerer Hämophilie erhielten 201 (68,4 %) eine prophylaktische Faktorensubstitution, während sich 84 (28,6 %) lediglich im Bedarfsfall mit einem entsprechenden Faktoren-Präparat behandelt wurden. Auffallend waren dabei die Unterschiede in der Altersverteilung – während bei Kindern und Jugendlichen (n = 87) bis 18 Jahre über 90 % mit einer regelmäßigen Faktoren-Prophylaxe behandelt wurden, lag der Anteil bei den über 50jährigen (n = 29) mit einer regelmäßigen Prophylaxe unter 40 %. Ältere Patienten mit schwerer Hämophilie, die bereits vor dem Bekanntwerden von Hepatitis und HIV in den 1980er-Jahren mit plasmatischen Produkten behandelt wurden, leiden weiterhin an diesen Folgen. So sind 113 (38,4 %) Patienten mit Hämophilie mit HCV infiziert und 37 (12,6 %) sind HIV-positiv.1 Die Erhebung der Behandlung der Patienten mit Hämophilie in Österreich zeigt die Herausforderungen der optimalen Therapie, insbesondere die Vermeidung hämophilieassoziierter Komplikationen und den Erhalt eines gesunden Gelenkstatus. Die bisher notwendigen regelmäßigen intravenösen Injektionen stellen zudem eine Belastung für den Patienten dar, zudem treten bei 30–40 % der Patienten zumindest vorübergehend im Laufe der Substitution Hemmkörper gegen den exogen zugeführten FVIII/IX auf.
Prophylaxe mit rekombinanten/plasmatischen Faktoren
Nach den infektionsbedingten Komplikationen (u. a. Hepatitis, HIV) zu Beginn der 1980er-Jahre kam es im Verlauf zu entsprechenden Sicherheitsauflagen für plasmatische FVIII/IX-Produkte, inkl. vorgeschriebener Virusinaktivierungsschritte, im Rahmen der Herstellung. Ende der 1990er-Jahre konnte FVIII/IX schlussendlich rekombinant in Zellkulturen hergestellt werden. Durch die somit auch steigende Verfügbarkeit von FVIII/IX-Präparaten wurde es möglich, Hämophilie-Patienten bei entsprechenden Kosten intensiver zu behandeln. Durch eine möglichst früh im Kindesalter begonnene, regelmäßige prophylaktische Faktorensubstitution – angepasst an das Körpergewicht und die patientenabhängigen Bedürfnisse – wurde versucht, Gelenksblutungen möglichst zu verhindern und die Entwicklung einer hämophilen Arthropathie zu vermeiden. Die individuell unterschiedlichen, jedoch generell kurzen Halbwertszeiten der FVIII/IX-Präparate machen jedoch, entsprechend den geltenden Richtlinien zur Vermeidung von spontanen Blutungen, regelmäßige intravenöse Gaben alle 2–3 Tage notwendig.2 Trotz der Verfügbarkeit der Produkte entscheiden sich weiterhin Patienten für eine Bedarfsbehandlung mit einer Substitution nur im Rahmen von besonderen Belastungen oder bei Blutungen, obwohl mehrfach gezeigt werden konnte, dass sich vermehrte Gelenkblutungen negativ auf den Gelenkstatus der Patienten auswirken.3 Seit der Einführung der rekombinanten FVIII-Präparate besteht auch die Diskussion, ob diese modifizierten Proteine zu einer erhöhten Hemmkörper-Entwicklung führen könnten. Mehrere kleinere Studien konnten diese Frage nicht eindeutig beantworten. Die bisher größte prospektive Studie zu diesem Thema („SIPPET Trial“; durchgeführt mit zuvor unbehandelten Kindern) zeigte eine geringere Rate von Hemmkörpern im Rahmen der Behandlung mit plasmatischen FVIII-Produkten im Vergleich zu rekombinanten FVIII-Präparaten.4
Produkte mit verlängerter Halbwertszeit
In den Jahren nach Einführung der ersten rekombinanten Faktor-Präparate kam es lediglich zu kleineren Modifikationen, speziell bei der Produktion der Präparate. Erst im Jahr 2016 kam es durch gentechnische und chemische Veränderungen der Gerinnungsfaktoren mittels PEGylierung bzw. Fusion mit einer Fc-Domäne oder Albumin zum nächsten großen Fortschritt in der Hämophilie-Behandlung.5 Speziell für entsprechende Faktor-IX-Präparate führten diese Modifikationen eine Verlängerung der Halbwertszeit um das bis zu 5-Fache auf über 100 Stunden. Die Substitutionsintervalle konnten bei weiterhin bestehendem Schutz vor Gelenksblutungen, angepasst an den Lebensstil des Patienten, auf bis zu 2 Wochen ausgeweitet werden. Die Interaktion mit dem von-Willebrand-Faktor hat zur Folge, dass die gleichen Modifikationen beim FVIII lediglich zu einer Verlängerung der Halbwertszeit um das ca. 1,5-Fache und somit im Schnitt auf 18–19 Stunden führen. Entsprechend den individuellen Bedürfnissen der Patienten können durch die Verwendung von FVIII-Präparaten mit verlängerter Halbwertszeit einerseits entweder unter Beibehaltung des ursprünglichen Substitutionsrhythmus höhere Talspiegel erreicht werden oder anderseits die Intervalle bis zur nächsten FVIII-Gabe verlängert werden.5 Im Rahmen des WFH-Meetings 2018 in Glasgow wurden erste Daten einer Phase-I/II-Studie von BIVV001 (rFVIIIFc-VWF-XTEN), einem rekombinanten FVIII-Molekül, vorgestellt, welches neben einer Fc-Domäne auch zusätzliche Anteile von VWF-Regionen und XTEN-Polypetiden aufweist. Die Halbwertszeit konnte durch diese Modifikationen auf 37 Stunden verlängert werden.
Faktor-VIII/IX-unabhängige Therapieansätze
Innovative Therapieansätze in der Behandlung der Hämophilie zielen, unabhängig von einer spezifischen Faktorensubstitution, auf ein neues Gleichgewicht in der Gerinnung ab. Bereits zugelassen in der Behandlung von Patienten mit einer Hämophilie A ist die Therapie mit Emicizumab (Hemlibra), einem chimären, bispezifischen, humanisierten monoklonalen Antikörper. Dieser verbindet den aktivierten FIX und den Gerinnungsfaktor X und ahmt dadurch die Funktion des fehlenden aktivierten FVIII nach. Die Wirksamkeit von Emicizumab ist zwar geringer als der FVIII6, jedoch kann Emicizumab subkutan verabreicht werden, die mittlere Halbwertszeit liegt bei 28 Tagen, und die Wirkung ist unabhängig von möglichen Hemmkörpern. Die bisher publizierten Zulassungsstudien im HAVEN-Studien-Programm zeigten prinzipiell eine sehr gute Wirksamkeit in der Vermeidung von Blutungen bei Kindern, Jugendlichen und Erwachsenen sowohl mit als auch ohne Hemmkörper.7, 8 Die Effektivität der Behandlung wurde primär gegen eine FVIII-Bedarfsbehandlung untersucht, ein direkter Vergleich mit adäquater FVIII-Substitution ist noch ausständig. Schwerwiegende Nebenwirkungen im Sinne von thromboembolischen Komplikationen und thrombotischen Mikroangiopathien traten lediglich im Rahmen einer gleichzeitigen Behandlung mit dem Bypassing-Medikament FEIBA auf. Die Rate der aufgetretenen Anti-Drug-Antibodys ist im Rahmen des bisher kurzen Beobachtungszeitraum niedrig.
Andere alternative Konzepte versuchen durch die Hemmung von endogenen antikoagulatorischen Faktoren wie z. B. Antithrombin und Tissue-Factor-Pathway-Inhibitor (TFPI) einen Ausgleich in der Gerinnungskaskade zu schaffen. Fitusiran, ein synthestisches siRNA-Oligonukleotid gegen Antithrombin III, oder Concizumab, ein anti-TFPI-Antikörper, zeigten in den publizierten Phase-I-Studien eine Normalisierung von Aktivierungsparametern in Hämophilie-Patienten.9, 10 Diese Medikamente sind subkutan sowohl bei Hämophilie A als auch B und unabhängig vom Hemmkörperstatus anwendbar. Langzeitergebnisse zur Sicherheit und Effektivität dieser Therapien bleiben abzuwarten.
Heilung der Erkrankung? – Gentherapie
Auch in der Gentherapie als mögliche potenzielle „Heilung“ der Erkrankung zeigen sehr rezent publizierte Daten erfreulicherweise sowohl in der Hämophilie A als auch Hämophilie B eine positive Entwicklung. Die einmalige Gabe eines mittels eines adenoassoziierten Virus-Serotype-5-(AAV5-)Vektors transferierten B-Domain-deletierten humanen FVIII (AAV5-hFVIII-SQ) führte in der Hochdosis-Gruppe bei 6 von 7 Patienten zu einer im Beobachtungszeitrum von 12 Monaten anhaltenden Faktor-VIII-Expression von über 50 IE/dl.11 Auch klinisch kam es unter der Therapie zu einem deutlichen Rückgang der Blutungsneigung. Die Nebenwirkungen der Therapie, insbesondere der Anstieg der Transaminasen, waren gut beherrschbar. In der Hämophilie B brachte der ebenfalls AAV-Vektor-vermittelte Gentransfer des Faktor IX mit einer Gain-of-Function-Mutation, FIX Padua („factor IX-R338L“), den möglichen Durchbruch.12 Bei 10 Patienten mit schwerer Hämophilie B konnte nach dem Gentransfer ein über den Beobachtungszeitraum von 28–78 Wochen anhaltender mittlerer FIX-Spiegel von 34 % (14–81 %) gemessen werden. Neun der Patienten hatten nach der Behandlung keine Blutungen mehr, und bei lediglich 2 der Patienten kam es vorübergehend zu einem Anstieg der Leberenzyme, welcher durch eine kurzzeitige Gabe von Prednisolon erfolgreich behandelt werden konnte.
Schlussfolgerungen
In den letzten Jahren wurden neue, innovative Therapieansätze für Patienten mit Hämophilie weiterentwickelt, welche zu einer deutlichen Reduktion der Belastung der Patienten führen können. Die Effektivität dieser Therapien im Vergleich zu einer adäquaten – angepasst an die individuellen Bedürfnisse der Patienten – Faktorensubstitution muss noch untersucht werden.
1 Retjö J et al., Hamostaseologie. 2018 Nov 12 [Epub ahead of print])
2 Pabinger I et al., Wikliwo 2015; 127 Suppl 3:S115–30
3 Nijdam A et al., Thromb Haemost 2016; 115(5):931–8
4 Peyvandi F et al., NEJM 2016; 374(21):2054–64
5 Mancuso ME et al., J Clin Med 2017; 6(4):39
6 Shima M et al., NEJM 2016; 26;374(21):2044–53
7 Oldenburg J. et al., N Engl J Med 2017; 377(9):809–818
8 Mahlangu J. et al., NEJM 2018; 379(9):811–822
9 Pasi KJ et al., NEJM 2017; 377(9):819–828
10 Chowdary P et al., J Thromb Haemost 2015; 13(5):743–54
11 Rangarajan S et al., NEJM 2017; 377(26):2519–2530
12 George LA et al., NEJM 2017; 377:2215–27

AutorIn: Dr. Florian Kocher
Universitätsklinik für Innere Medizin V – Hämatologie und Onkologie,Medizinische Universität Innsbruck

AutorIn: Ass. Prof. Priv.-Doz. Dr. Clemens Feistritzer
Universitätsklinik für Innere Medizin V – Hämatologie und Onkologie,Medizinische Universität Innsbruck

Ursprünglich erschienen:
UIM 04|2019
UIM 04|2019
