


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Rheumatische Differenzialdiagnose – Muskelschmerz
28. November 2018
Muskuloskelettale Schmerzen gehören zu den häufigsten Beschwerden in der Bevölkerung und sind in Österreich für nahezu jeden 10. Krankenstand und für 22 % aller Invaliditätspensionen verantwortlich. Eine entsprechende Früherkennung und Behandlung dieser Beschwerden ist daher nicht nur aus Sicht der Patienten, sondern auch aus gesundheitsökonomischer Sicht von großer Bedeutung.
Fokale und diffuse Myalgien
Verschiedenste Krankheiten können Grund für Muskelschmerzen sein, was die Suche nach der Ursache zu einem schwierigen Unterfangen macht. Wichtig ist daher eine zielgerichtete Anamnese, in der nach Lokalisation, Stärke, Beginn, Dauer und Umstände, unter denen die Schmerzen auftreten, gefragt wird.
Grundlegend kann man zwischen fokalen Myalgien, die einzelne Muskeln betreffen, und diffusen Myalgien, bei denen verschiedene Muskelgruppen betroffen sind, unterscheiden.
Fokale Myalgien, die durch repetitive Bewegungen verstärkt und durch Relaxation gebessert werden, gehen meist auf Überbeanspruchung der entsprechenden Struktur zurück. Es können jedoch auch neuromotorische Erkrankungen oder Gefäßverschlüsse die Ursache für fokale Myalgien sein, weshalb Neuro- und Gefäßstatus bei der Untersuchung von Muskelschmerzen immer mitgemacht werden sollten.
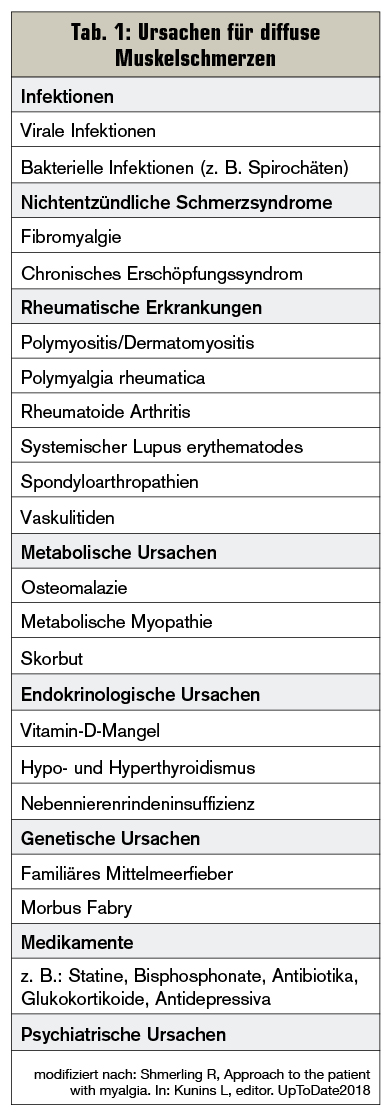
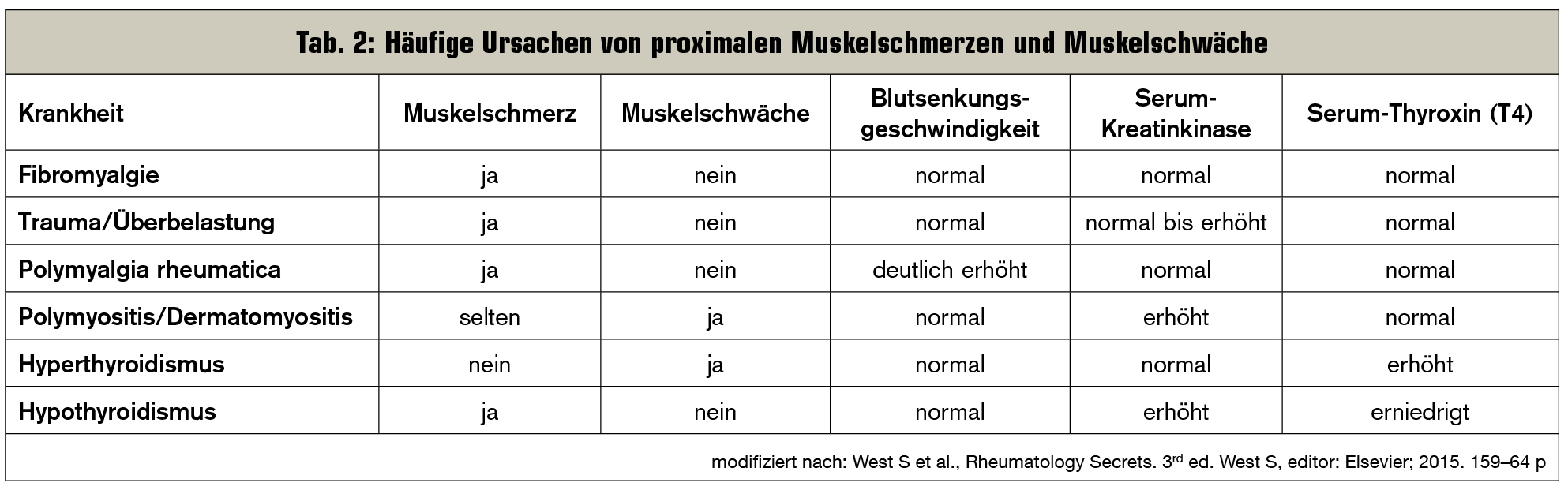
Diffuse Myalgien hingegen können infektiöse, endokrinologische, metabolische, genetische, medikamenteninduzierte, aber auch rheumatische Ursachen haben (Tab. 1), wobei häufig die proximalen Muskelgruppen betroffen sind (Tab. 2):
- Bei der Polymyalgia rheumatica klagen Patienten (&re;= 50 Jahre) über subakut einsetzende Schmerzen und Morgensteifigkeit im Bereich der Schultern und der Hüften. Im Labor zeigt sich meist eine hohe Blutsenkungsgeschwindigkeit, und eine Therapie mit Glukokortikoiden führt in der Regel zu einer schnellen Verbesserung der Beschwerden.
- Fibromyalgie ist ein chronisches, nichtentzündliches Schmerzverarbeitungssyndrom, bei dem Patienten Schmerzen im Bereich des Muskel–Sehnen-Übergangs verspüren. Muskelenzyme und Entzündungsparameter sind unauffällig.
- Weitere Ursachen für diffuse Myalgien können statininduzierte Myopathien sein, aber auch das zu schnelle Absetzen von Antidepressiva kann zu starken Muskelschmerzen führen.
- Um Hyper- oder Hypothyroidismus als Ursachen ausschließen zu können, sollten auch stets Schilddrüsenhormone bei diffusen proximalen Muskelschmerzen überprüft werden.
Idiopathisch entzündliche Myopathien
Eine weitere wichtige Gruppe im Kontext der diffusen Myopathien stellen die idiopathischen entzündlichen Muskelerkrankungen dar, zu denen die adulte Polymyositis und die Dermatomyositis, die Einschlusskörpermyositis und die juvenile Dermatomyositis gehören.
Es handelt sich generell um seltene Erkrankungen mit einer Inzidenz von 1 : 100.000 Fälle pro Jahr, wobei Frauen dreimal häufiger erkranken als Männer. Bei der juvenilen Dermatomyositis ist das durchschnittliche Alter bei Beginn der ersten Symptome 5–15 Jahre, während die adulte Form der Dermatomyositis sowie die Polymyositis normalerweise zwischen dem 50. und 60. Lebensjahr auftreten.
Das Leitsymptom dieser Krankheiten ist die proximale Muskelschwäche. Betroffene berichten über seit Monaten zunehmende Schwäche der Schulter- und Hüftmuskeln. Alltägliche Tätigkeiten, wie das Stiegensteigen, das Tragen von schweren Einkäufen oder das Kämmen der Haare, bereiten immer größere Schwierigkeiten. Es können außerdem auch die Muskeln des Pharynx und des Ösophagus betroffen sein, was zu Dysphonie und Dysphagie führen kann. Bei Poly- und Dermatomyositis ist die proximale Muskelschwäche bilateral, während sie bei der Einschlusskörpermyositis einseitig sein kann und auch bis in die distale Muskulatur reichen kann.
Interessanterweise geben Patienten bei diesen Krankheiten nur selten Muskelschmerzen (< 25 %) an, die – wenn überhaupt – nur als schwach empfunden werden (Tab. 2).
Bei der Dermatomyositis finden sich außerdem typische Hautveränderungen, wie das heliotrope Erythem (weinrote Erythme im Bereich der Augenlider), Gottron-Papeln (sattrote, streifige Papeln an der Fingerstreckseite) oder die periorale Blässe.
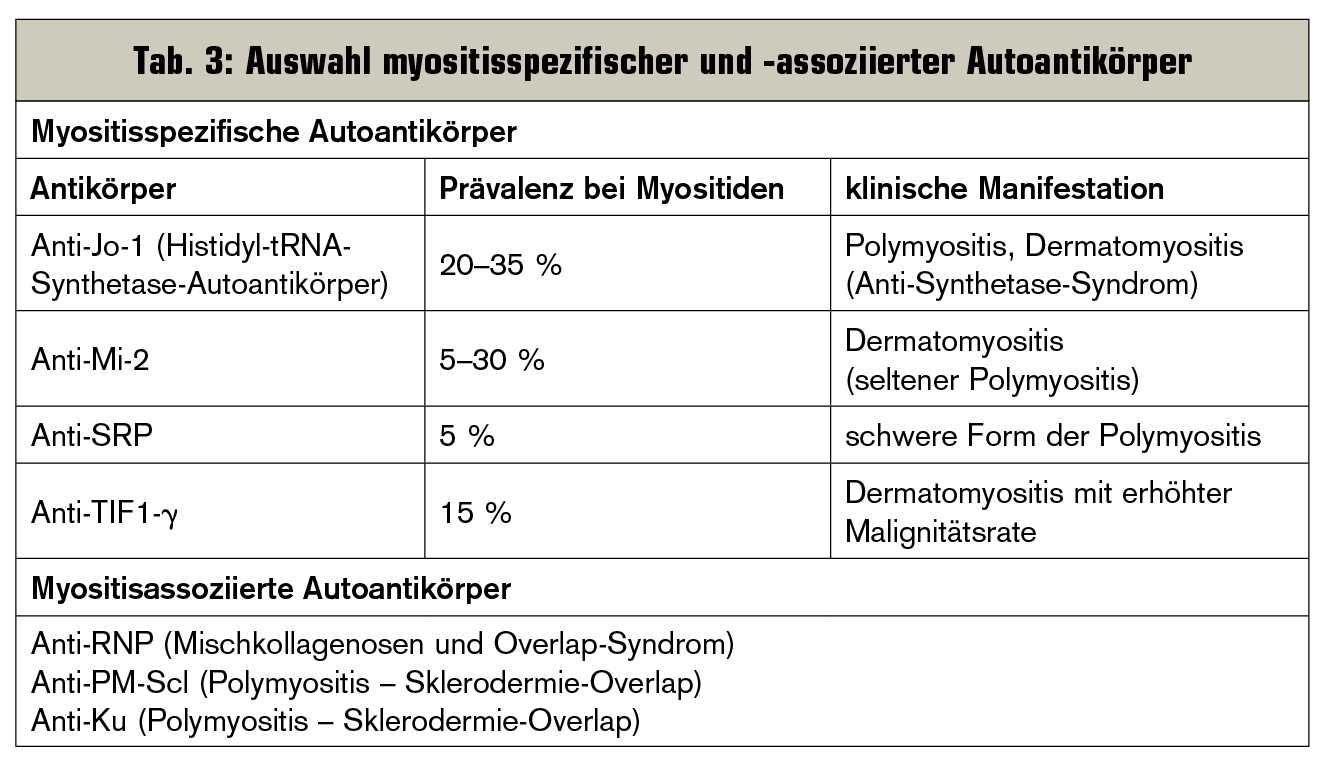
Um die Verdachtsdiagnose zu erhärten, werden im Serum Leitenzyme wie die Kreatinkinase, Aldolase, Myoglobin sowie Leberfermente und die Laktatdehydrogenase bestimmt, die bei diesen Krankheiten zumeist erhöht sind. Es kann außerdem eine Myoglobinurie vorliegen. Myositisspezifische Autoantikörper (Tab. 3), wie Anti-Jo-1, Anti-SRP oder Anti-Mi-2, kommen bei bis zu 50 % der Patienten mit idiopathisch entzündlichen Myopathien vor und sind neben einer diagnostischen Hilfe auch Prognosefaktoren für die Schwere des Krankheitsverlaufs. Myositisassoziierte Autoantikörper sind häufig auch erhöht, haben aber eine geringere Spezifität für Myositiden und kommen auch bei anderen rheumatischen Erkrankungen und Overlap-Syndromen (z. B. Mischung aus Polymyosits und Sklerodermie) vor.
Im Immunlabor der Rheumaabteilung an der Medizinischen Universität Graz werden zusätzlich bei Verdacht auf Autoimmunmyositis die Antikörper gegen OJ, EJ, PL-12, PL-7, PM-SCL100, SAE1, NXP2 und MDA5 bestimmt. Eine weitere Hilfe ist die Untersuchung mittels Elektromyogramm, welches bei Polymyositis und Dermatomyosits zwar eine hohe Sensitivität (85 %), aber eine niedrige Spezifität (33 %) aufweist. Eine Muskelbiopsie kann diagnostische und prognostische Informationen liefern und ist notwendig, um die „rot umrandeten Vakuolen mit β-Amyloid“ darzustellen, die sich bei der Einschlusskörpermyositis finden. Seit 2017 existieren neue Klassifizierungskriterien für die entzündlich idiopathischen Muskelerkrankungen, welche zwar in erster Linie für die Klassifizierung von Studienpatienten gestaltet wurden, aber auch im klinischen Alltag als Entscheidungsstütze genutzt werden können.
Das primäre Behandlungsziel bei der Poly- und Dermatomyositis ist das „Einbremsen“ des eigenen Immunsystems, um weitere Muskelschäden zu minimieren. Dafür erfolgt eine initiale Therapie mit Glukokortikoiden (0,75 bis 1 mg/kg Körpergewicht Prednisolonäquivalent), die nach 3–4 Wochen, unter Kontrolle der Muskelenzyme, langsam reduziert werden sollte. Weitere immunsuppressive Therapien, wie Methotrexat, Azathioprin, Ciclosporin, Tacrolimus, Rituximab etc., können zusätzlich verwendet werden, um die Menge an Glukokortikoiden möglichst niedrig zu halten. Körperliche Betätigung und Physiotherapie sind, auch bei aktiver Entzündung, wichtige Therapieansätze für Patienten mit Myositiden.
Adulte Poly- und Dermatomyositis (vor allem bei vorhandenem Autoantikörper Anti-TIF1-γ) gehen außerdem mit einem bis zu 6-fach erhöhten Risiko für maligne Erkrankungen einher. Da verschiedene Organsysteme betroffen sein können (Pankreas, Kolon, Brust, Ovarien etc.), ist ein Tumorscreening vor allem in den ersten drei Jahren nach Symptombeginn (in diesem Zeitraum ist das Risiko am höchsten) essenziell.
Resümee: Ob fokale oder diffuse Muskelschmerzen – eine genaue Anamnese und körperliche Untersuchung sind unerlässlich für die Ursachenfindung und Diagnosesicherung. Idiopathisch entzündliche Myositiden, wie die Dermatomyositis und Polymyositis, äußern sich weniger durch Muskelschmerzen, als viel mehr durch eine proximale, bilaterale Muskelschwäche. Sollte der begründete Verdacht einer rheumatischen Muskelerkrankung bestehen, ist die Involvierung eines Rheumatologen frühzeitig von Vorteil, um mögliche Spätfolgen zu verhindern.

AutorIn: Dr. Philipp Bosch
Klinische Abteilung für Rheumatologie und Immunologie,Universitätsklinik für Innere Medizin, Medizinische Universität Graz

Ursprünglich erschienen:
UIM 09|2018
UIM 09|2018