


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Schwere neonatale Thrombosen bei homozygotem Antithrombinmangel
28. November 2018
Einleitung: Wir berichten über vier Kinder aus verschiedenen Familien mit homozygotem Antithrombinmangel Typ II, einer seltenen, angeborenen Thrombophilie. Alle Kinder entwickelten kurz nach der Geburt spontane schwere venöse und/oder arterielle Thrombosen.1
Hintergrund
Thrombosen bei Neugeborenen sind selten und treten meist als sekundäre Komplikation von Grunderkrankungen auf, wie einer Sepsis oder eines angeborenen Herzfehlers, oder exogenen Triggern wie intravaskulären Kathetern. Hingegen spielen endogene prothrombotische Risikofaktoren im Kindesalter selten eine Rolle bei der Entstehung von Thrombosen, während bei Erwachsenen angeborene Thrombophilien das Thromboserisiko erhöhen.
Antithrombin (AT) ist ein wichtiger physiologischer Hemmer von Gerinnungsfaktoren, vor allem von Faktor Xa und Thrombin. Ein AT-Mangel erhöht das Risiko von venösen und arteriellen Thrombosen. Es werden zwei Formen des angeborenen AT-Mangels beschrieben: der quantitative Mangel (Typ I) mit niedrigem AT-Antigen und AT-Aktivität, sowie der qualitative Mangel (Typ II) mit einem dysfunktionalen Protein, definiert durch einen annähernd normalen AT-Antigenspiegel, aber reduzierter AT-Aktivität.
Während der AT-Mangel Typ I in homozygoter Form nicht mit dem Leben vereinbar ist, dürfte der mildere AT-Mangel Typ II in homozygoter Form lebensfähig sein, wie einzelne Fälle in der Literatur illustrieren. Wir berichten über vier Neugeborene mit homozygotem AT-Mangel Typ II, bei denen spontane schwere Thrombosen in den ersten Lebenswochen auftraten.
Fallberichte
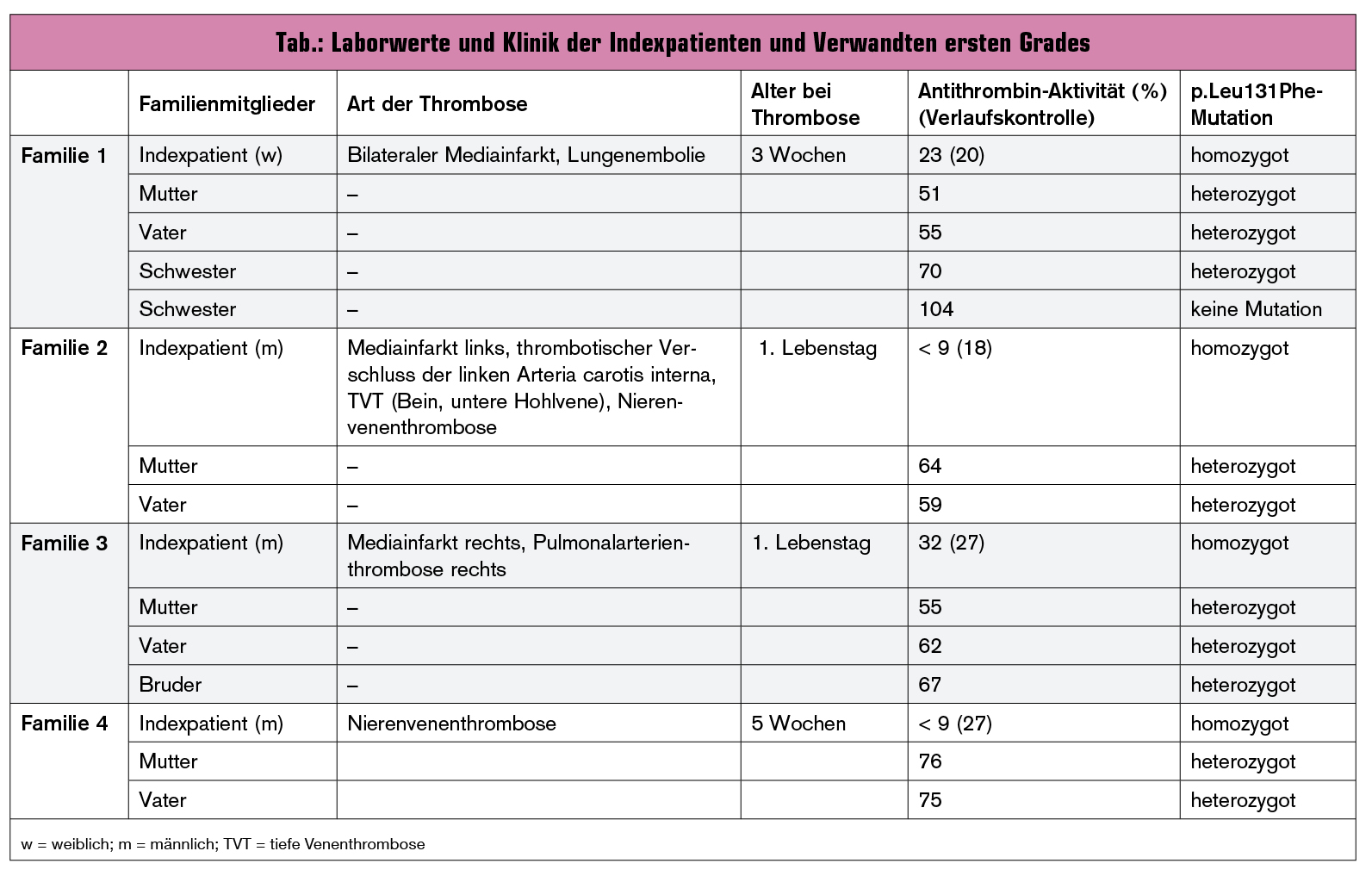
Klinische Präsentation: Die Patienten präsentierten sich im Alter von einem Tag bis zu fünf Wochen mit spontanen Thrombosen (Tab.). Diese umfassten eine akute Lungenembolie mit Kreislaufversagen im Alter von drei Wochen wie bei Patientin 1, bilateralen und unilateralen Mediainfarkt (Patient 1, 2 und 3), Beinvenenthrombose (Patient 2 und 3) sowie Nierenvenenthrombose (Patient 2 und 4). Die Diagnose der Thrombose war teilweise durch Komorbiditäten erschwert, wie bei Patient 4, einem Frühgeborenen in der 28. Schwangerschaftswoche.
Familienanamnese: Alle vier Patienten sind Kinder von klinisch gesunden, nicht konsanguinen Eltern und haben gesunde Geschwister. In der Familienanamnese findet sich keine Thrombose der Verwandten ersten Grades. Die Eltern von Patient 3 berichteten über mehrere Thrombosen in der Familie väterlicherseits (Großmutter, zwei Großtanten und ein Urgroßvater). Alle Familien stammen aus verschiedenen Regionen in Serbien, die Großeltern von Patient 3 und 4 sind Roma.
Diagnose des Antithrombinmangels: Die ersten, nach klinischer Diagnose der Thrombosen, gemessenen AT-Aktivitäten der vier Kinder betrugen 23 %, 32 % und < 9 % (Tab.). Diese Werte führten jedoch nicht immer gleich zur Diagnose eines AT-Mangels, da sie auch durch das junge Alter und den kritischen Zustandes der Patienten erklärbar waren; bei Patient 4 war zudem die Untersuchung der Eltern unauffällig (AT-Aktivität > 70 %).
In allen Familien konnte die DNA-Analyse die Diagnose eines AT-Mangels Typ II, bedingt durch die p.Leu131Phe-Missense-Mutation (auch p.Leu99Phe oder Budapest III [ATBp3]), bestätigen. Die Mutation war homozygot bei allen vier Indexpatienten und heterozygot bei allen Eltern und betroffenen Geschwistern. Bei keinem der Patienten konnten weitere Thrombophilien nachgewiesen werden.
Therapie: Die Mutation, die zu einem Defekt der Heparinbindungsstelle führt, bedingte ein inadäquates Ansprechen auf Heparin. Bei allen Patienten konnten nur unter wiederholter Substitution mit AT-Konzentrat therapeutische Anti-Xa-Zielwerte erreicht werden.
Bei den Patienten 2 und 4 wurden Dosierungen von 53–63 IU/kg AT-Konzentrat täglich und Enoxaparin mit 2 mg/kg bzw. 2,3 mg/kg zweimal täglich verabreicht, um therapeutische Anti-Xa-Spiegel zu erzielen. Die Enoxaparin-Dosen waren höher als die in diesem Alter empfohlene Dosis von 1,5 mg/kg, jedoch im Einklang mit Dosierungen bei kritisch kranken Neugeborenen. Bei Patient 3 wurde der direkte Thrombininhibitor Bivalirudin mit Dosierungen bis zu 0,125 mg/kg/h verabreicht. Der Patient entwickelte jedoch im Verlauf eine katheterassoziierte Thrombose im Bereich der Oberschenkel- und Beckenvenen. Diese Komplikation könnte durch eine inadäquate Bivalirudin-Dosis bedingt gewesen sein, da es keine altersentsprechenden Dosierungsempfehlungen gibt.
Alle Kinder wurden in den ersten vier Lebensmonaten auf eine Therapie mit Vitamin-K-Antagonisten (VKA) umgestellt. In diesem Alter ist der Dosisbedarf für VKA aufgrund des raschen Wachstums, niedrigen Vitamin-K-Gehaltes der Muttermilch bzw. hohen Vitamin-K-Gehaltes von Formula-Nahrungen sehr variabel, was häufige INR-Messungen erfordert.
Während einer Nachbeobachtungszeit von 12 Jahren, 8 Jahren, 4,5 Jahren bzw. 2 Jahren kam es bei keinem der Kinder zu einer erneuten Thrombose noch zu Blutungskomplikationen unter der VKA-Therapie.
Diskussion
Bei allen vier Patienten manifestierten sich schwere Thrombosen in den ersten Lebenswochen. Nach klinischem Verdacht konnte ein homozygoter AT-Mangel Typ II, bedingt durch die p.Leu131Phe-Mutation, diagnostiziert werden. Im Falle einer Thrombose beim Neugeborenen ohne ersichtlichen klinischen Auslöser sollte an schwere Thrombophilien, wie homozygoten AT-, Protein-C- oder Protein-S-Mangel gedacht werden.
Der empfohlene initiale Test zur Diagnose eines AT-Mangels ist ein funktionaler AT-Assay (z. B. Anti-IIa-Heparin-Kofaktor-AT-Assay), da AT-Antigen bei AT-Mangel Typ II im Normalbereich liegen kann. Überdies kann der AT-Spiegel auch als Folge einer Thrombose oder initial während einer Heparintherapie verringert sein. Auch sind bei gesunden Neugeborenen und Frühgeborenen die AT-Spiegel physiologisch niedriger und gleichen sich erst im Alter von etwa sechs Monaten jenen von Erwachsenen an. Sobald ein angeborener AT-Mangel vermutet wird, sollten auch die Eltern getestet werden. Die genetische Untersuchung erbringt die definitive Diagnose.
Eine frühe Diagnose des AT-Mangels ist zur Einleitung der richtigen Therapie wichtig. Die Standard-Antikoagulation mit Heparin ist bei AT-Mangel potenziell ineffektiv. Für eine wirksame Heparintherapie ist meist die zusätzliche Substitution von AT-Konzentrat notwendig. Die Dosisfindung für Anti-Xa-Zielwerte im therapeutischen Bereich kann einige Tage dauern, besonders in der Neugeborenenperiode. Das Unvermögen, adäquate Anti-Xa-Zielwerte unter Heparin zu erzielen, kann ein erster Hinweis auf einen AT-Mangel sein. Langfristig ist die Antikoagulation mit VKA die Therapie der Wahl. Die Behandlung mit direkten Gerinnungshemmern könnte eine Alternative darstellen, sobald altersentsprechende Dosierungen für Säuglinge und Kinder etabliert sind. Bei Patienten mit homozygotem AT-Mangel muss aufgrund des hohen Thromboserisikos eine lebenslange Antikoagulation empfohlen werden.
Resümee
Im Falle von Thrombosen beim Neugeborenen ohne klinischen Auslöser sollte an schwere Thrombophilien, wie homozygoter AT-, Protein-C- oder Protein-S-Mangel, gedacht werden. Der seltene homozygote AT-Mangel-Typ II kann auch ohne Thromboseanamnese der Eltern auftreten. Bei schwerem AT-Mangel ist eine Heparintherapie meist nur in Kombination mit Substitution von AT-Konzentrat wirksam, und es ist eine frühe Umstellung auf alternative Gerinnungshemmer, wie VKA, erforderlich.
1 Swoboda V et al., Thrombosis Research 2017; 158:134–137

AutorIn: Dr. Vanessa Swoboda
Klinische Abteilung für Pädiatrische Kardiologie, Universitätsklinik für Kinder- und Jugendheilkunde, Medizinische Universität Wien

AutorIn: OÄ Dr. Katharina Thom
Klinische Abteilung für Pädiatrische Kardiologie, Universitätsklinik für Kinder- und Jugendheilkunde, Medizinische Universität Wien

AutorIn: AO Univ.-Prof. Dr. Christoph Male
Klinische Abteilung für Pädiatrische Kardiologie, Universitätsklinik für Kinder- und Jugendheilkunde, Medizinische Universität Wien

Ursprünglich erschienen:
UIM 09|2018
UIM 09|2018
