
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Update 2021 – Hämoglobinopathien
29. März 2021
Unter „Hämoglobinopathien“ versteht man Erkrankungen, welche den Blutfarbstoff Hämoglobin betreffen. Sie verursachen nur dann eine Krankheit oder hämatologische Veränderungen, wenn der molekulare Fehler mit einer Störung der Funktion, Löslichkeit, Stabilität oder mit einer verminderten Synthese der Hämoglobinstruktur einhergeht. Daraus lässt sich auch erklären, warum für die Klinik nur wenige Hämoglobinopathien wirklich klinisch relevant sind, obwohl mittlerweile eine enorme Anzahl an verschiedenen Hämoglobinvarianten bekannt sind.1 Das erste anomale Hämoglobin wurde 1940 vom zweifachen Nobelpreisträger Linus Pauling beschrieben. Er entdeckte bei Sichelzellerkrankten eine veränderte Hämoglobinform der Erythrozyten. Sechs Jahre später schaffte er es, die molekularbiologischen Abweichungen des Sichelzellhämoglobins zu beschreiben.2 Durch seine Errungenschaften kurbelte Pauling das Interesse vieler Forscher an und verhalf der molekularen Medizin zu ihrer Geburt.
Aufbau: Hämoglobin umfasst vier Untereinheiten mit jeweils einer Polypeptidkette und einer Hämgruppe. Es gibt zwei Arten von Polypeptidketten des adulten Hämoglobins, die als Alpha- und Betaketten bekannt sind. Sie sind ähnlich lang, unterscheiden sich jedoch in der Aminosäurensequenz. Die Alpha-Kette aller menschlichen Hämoglobine, fetal oder adult, ist dieselbe. Die Nicht-Alpha-Ketten umfassen die Beta-Kette von normalem adultem Hämoglobin (α2β2), die Gamma-Kette von fetalem Hämoglobin (α2γ2) und die Delta-Kette von HbA2. Die Struktur von Hämoglobin wurde ausführlich durch Röntgenanalysen untersucht. Die Anordnung der Untereinheiten, die als quaternäre Struktur bekannt ist, unterscheidet sich im Oxy- und Desoxyhämoglobin.3 Fetales Hämoglobin (HbF) ist die dominante Form von Hämoglobin, die während der Schwangerschaft im Fötus vorhanden ist. HbF wird von erythroiden Vorläuferzellen in der 10. bis 12. Schwangerschaftswoche und in den ersten sechs Monaten des postnatalen Lebens produziert. Im Gegensatz zum adulten Hämoglobin enthält HbF zwei Alpha- und zwei Gamma-Untereinheiten. Im Alter von 6 bis 12 Monaten wird fast das gesamte fetale Hämoglobin durch HbA ersetzt, es sei denn, beim Individuum liegt eine Hämoglobinopathie vor. Beim Erwachsenen liegt der Restanteil an HbF in der Regel unter 0,5 % des Gesamthämoglobins. Der Anteil an fetalem Hämoglobin ist nämlich für die Diagnostik und Pathophysiologie von Hämoglobinopathien von Relevanz.4
Hämoglobinvarianten
Einteilung: Die Hämoglobinvarianten umfassen alle genetischen Erkrankungen des Hämoglobins und können Störungen funktioneller und struktureller Art zur Folge haben. Grob kann man sie in drei Hauptgruppen unterteilen: Erstens kann eine reduzierte Synthese von normalen Globinketten vorliegen, welche den Thalassämien zugeordnet werden. Zweitens sind auch strukturelle Defekte des Hämoglobins bekannt. Überschneidungen zwischen beiden sind auch möglich, sodass ein Patient eine verminderte Bildung eines abnormalen Hämoglobins aufweisen kann, welche sich dann aber vom Phänotyp her wie eine Thalassämie präsentiert. Des Weiteren gibt es Störungen bei der Umstellung der Bildung von fetalem zu adultem Hämoglobin, die auch als „hereditäre Persistenz“ von fetalem Hämoglobin bezeichnet wird.5
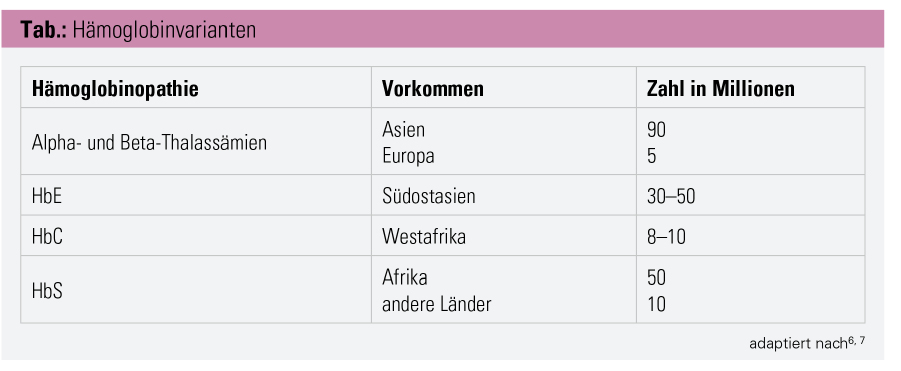
Epidemiologie: Weltweit sind 7 % der Bevölkerung von Hämoglobinopathien betroffen. Damit gehören sie zu den häufigsten Erbkrankheiten. In Nord- und Mitteleuropa, einschließlich Österreich, wurden sie in letzter Zeit aufgrund von Einwanderung viel häufiger. Hämoglobinopathien sind in der heutigen multiethnischen europäischen Bevölkerung ein Problem der öffentlichen Gesundheit. Eine angemessene Versorgung der betroffenen Patienten erfordert eine Vielzahl diagnostischer und therapeutischer Maßnahmen.6 Hämoglobinvarianten sind folgendermaßen aufgeteilt (siehe Tab.): Alpha- und Beta-Thalassämien findet man überwiegend in Asien und Europa, vor allem im Mittelmeerraum. Die Sichelzellanämie hat ihren Ursprung in Afrika, während das HbC vor allem in Westafrika aufzufinden ist. HbE ist besonders in Südostasien verbreitet.6, 7
Seit 1998 existiert auch eine elektronische Datenbank zu Hämoglobinvarianten, auffindbar unter http://globin.cse.psu.edu, wo alle bisher über 700 entschlüsselten Hämoglobinvarianten aufgelistet werden. Erwähnenswert ist, dass in denselben geographischen Regionen genauso gehäuft auch der Glukose-6-Phosphat-Dehydrogenase-Mangel vorkommt, der unter anderem auch der häufigste bekannte menschliche Enzymdefekt ist. Der Enzymmangel betrifft weltweit mehr als 400 Millionen Menschen und kann, ausgelöst durch verschiedene Substanzen inklusive Medikamente, zu hämolytischen Anämien führen.8
Molekulare Grundlagen: Anomale Hämoglobine sind durch einen Defekt in der Primärstruktur des Globins gekennzeichnet, also durch einen Fehler in der Sequenz der Aminosäuren der Polypeptidkette. Die Ursache der Hämoglobinvarianten sind Defekte in der Synthese des Globinmoleküls durch Mutationen im genetischen Code. Solche Punktmutationen stellen die häufigsten Varianten wie HbC oder HbS dar. Hämoglobintests sind insbesondere in den folgenden Situationen angezeigt:6, 7
- mikrozytäre Anämie, nachdem ein Eisenmangel ausgeschlossen bzw. behoben wurde.
- chronische hämolytische Anämie
- Gefäßverschlusskrisen unklarer Ätiologie bei Patienten aus Gebieten, in denen HbS und/oder HbC weitverbreitet sind.
- medikamenteninduzierte Anämien
- Erythrozytosen und/oder Zyanosen, die durch hämatologische Faktoren ausgelöst werden können.
- Hydrops fetalis unklarer Genese
- Prävention (Testung von Familienmitgliedern, Diagnostik von Partnern bei der genetischen Beratung)
- pränatale Diagnostik: alle Schwangeren aus Risikopopulationen, im positiven Fall auch der Partner
Diagnostik
Die Diagnose in der Routinepraxis umfasst die Anzahl der Erythrozyten (RBC) mit Erythrozytenindizes (MCV, MCH, MCHC), einen Hämoglobintest sowie Hämoglobin-Elektrophorese (Abb. 1) und/oder Chromatografie.6 Der Mentzer-Index, der sich aus dem Quotienten von MCV und Erythrozyten ableitet, ist mit einer Sensitivität von 98,7 % und einer Spezifität von 82,3 % der verlässlichste Index zur Differenzierung zwischen Beta-Thalassämie und Eisenmangel.8 Ein Mentzer-Index > 13 spricht für eine Eisenmangelanämie, während ein Index < 13 auf eine Beta-Thalassämie hinweist. Ein Eisenmangel sollte grundsätzlich vor der Diagnostik einer Hämoglobinopathie ausgeschlossen und substituiert werden, da dieser z. B. eine Beta-Thalassämie maskieren kann. Zusätzlich sind weitere labormedizinische Analysen hilfreich, etwa die Bestimmung von Ferritin und der Transferrrinsättigung, Hämolyseparameter (Haptoglobin, Hämopexin, LDH, Bilirubin), Antikörpersuchtest und Leberfunktionsparameter (GOT, GPT, γGT, Cholinesterase).6 Um auch seltenere Hämoglobinopathien oder Compound-Heterozytogien (z. B. HbS/β0-Thal) zu erfassen, kann eine weitere molekularbiologische Charakterisierung die Diagnose sichern.9

Klinik
Abgesehen von asymptomatischen Trägern können sich Hämoglobinopathien klinisch als persistierende Anämie, Hämolyse, Polyglobulie oder Zyanosen unklarer Genese bis hin zu schweren Sichelzellkrisen mit Beschwerden im Rücken, in den Extremitäten und im Thoraxbereich sowie mit Bauchschmerzen oder auch neurologischen Symptomen präsentieren. Patienten sind zudem anfälliger für Infektionen, insbesondere durch Pneumokokken, Hämophilus, Salmonellen, Klebsiellae und Mykoplasmen.6 Die Stärke der Ausprägung von Symptomen ist neben weiteren Faktoren zusätzlich davon abhängig, ob ein Patient heterozygoter oder homozygoter Träger einer Anomalie ist.10 Oft fallen Hämoglobinvarianten erstmalig als Zufallsbefund im Labor im Rahmen der HbA1c-Bestimmung auf. Die klassi
sche Bestimmung des HbA1c-Wertes erfolgt mittels Hoch-leistungsflüssigkeitschromatografie (HPLC; Abb. 2). Sie ist auch eine Methode, um normales von abnormem Hämoglobin zu trennen. Im Rahmen der Diagnostik von Thalassämien können damit genaue reproduzierbare HbA2-Werte ermittelt werden. Liegt eine Hämoglobinopathie vor, ist die Interpretation des HbA1c-Wertes mit Limitationen bzw. Einschränkungen verbunden. Hier bieten sich alternativ andere Marker zur Kontrolle des Langzeitzuckers (z. B. Fruktosamin) an; allerdings dürfen diese nicht zur Diagnosesicherung eines Diabetes herangezogen werden.11

Therapie
Verschiedene Hämoglobinopathien erfordern entweder keine oder unterschiedliche Therapien. Diese reichen von symptomatischen Therapien bis hin zu regelmäßigen Bluttransfusionen, zur Verwendung von Eisenchelatoren und zur Transplantation von hämatopoietischen Stammzellen. Derzeitige Forschungsprojekte fokussieren auf neue Ansatzmöglichkeiten, um die Erkrankungen mittels Gentherapie mit viralen Vektoren oder Designer-Nuklease-basierter Gen-Bearbeitung zu heilen.6, 12, 13
Protektive Wirkungen: Der Schutz vor schweren Malariasyndromen ist für die Sichelzellerkrankung sowie für HbCC, HbAC sowie homozygote und heterozygote α-Thalassämie-Träger von Bedeutung. Diese Hämoglobinopathien unterscheiden sich jedoch erheblich im Schutzgrad.14 Aufgrund eines selektiven Vorteils in Malariaregionen sind diese Hämoglobindefekte besonders häufig in Afrika, Asien und im Mittelmeerraum sowie bei Patienten, die aus den entsprechenden Gebieten zu uns herzogen.15
Fazit
Die Lebensdauer und vor allem die Lebensqualität von Patienten mit schweren Hämoglobinstörungen können mit heutigen Behandlungsmethoden signifikant verbessert werden. Umfassende Ansätze zur Diagnosesicherung und nachfolgende Behandlungsstrategien lassen sich noch optimieren. Ein Hauptaugenmerk sollte auch auf die Fortsetzung der Behandlung und Betreuung solcher Patienten gelegt werden, die vom jugendlichen Alter ins Erwachsenenalter übergingen.6 Sowohl Kinder als auch Erwachsene mit bekannter Sichelzellhomozytogie sollten beim Auftreten von Schmerzen klinisch ernstgenommen werden, da es sich um eine Sichelzellkrise handeln kann, die mit einer erhöhten Morbidität und Mortalität assoziiert ist.16
- Huber AR et al., Swiss Med Forum 2004; 4:921–6
- Eaton WA, Am J Hematol 2020; 95(2):205–11
- Marengo-Rowe AJ, Proc (Bayl Univ Med Cent) 2006; 19(3):239–45
- Steinberg MH, J Clin Med 2020; 9(11):3782
- Forget BG, Ann N Y Acad Sci 1998; 850:38–44
- Kohne E, Dtsch Arztebl Int 2011; 108(31–32):532–40
- Kohne E, Dtsch Arztebl Int 2010; 107(5):65–71
- Nkhoma ET et al., Blood Cells Mol Dis 2009; 42(3):267–78
- Vehapoglu A et al., Anemia 2014; Article ID 576738, DOI:10.1155/2014/576738
- Houwing ME et al., Blood Rev 2019; 37:100580
- Kohnert KD et al., World J Diabetes 2015; 6(1):17–29
- Traeger-Synodinos J et al., Eur J Hum Genet 2015; 23(4):426–37
- Brendel C et al., Curr Opin Hematol 2020; 27(3):149–54
- Taylor SM et al., Lancet Infect Dis 2013; 12(6):457–68
- Iolascon A et al., Hemasphere 2019; 3(3):e208
- Novelli EM, Gladwin MT, Chest 2016; 149(4):1082–93

AutorIn: Dr. Vilmos Fux
Zentralinstitut für med. u. chem. Labordiagnostik (ZIMCL)
LKH-Universitätskliniken Innsbruck

Ursprünglich erschienen:
UIM 02|2021
UIM 02|2021
