


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Virtuelle Updates für die klinische Praxis
24. Juli 2020
Neu diagnostizierte AML: neue Therapieoption bei älteren Patienten
Patienten mit akuter myeloischer Leukämie (AML), die nicht für eine Intensivtherapie in Betracht kommen, wiesen unter bislang verfügbaren Therapien geringe Ansprechraten und eine ungünstige Prognose auf.
Methoden: Die randomisierte, doppelblinde Phase-III-Studie VIALE-A evaluierte die Kombinationstherapie mit Venetoclax (VEN) und Azacitidin (AZA) gegenüber AZA allein in diesem unterversorgten Patientenkollektiv. 431 AML-Patienten, die aufgrund von Komorbiditäten und/oder höherem Alter ≥ 75 Jahre (medianes Studienalter 76 Jahre) keine intensive Induktionstherapie erhalten konnten, wurden 2 : 1 randomisiert mit VEN + AZA bzw. PBO + AZA behandelt; primärer Endpunkt war das Gesamtüberleben (OS).
Ergebnisse: Nach einem medianen Follow-up von 20,5 Monaten lag das mediane OS in der VEN+AZA-Gruppe bei 14,7 Monaten vs. 9,6 Monate unter PBO + AZA, was einer statistisch signifikanten Reduktion des Mortalitätsrisikos um 34 % entspricht (Abb. 1).
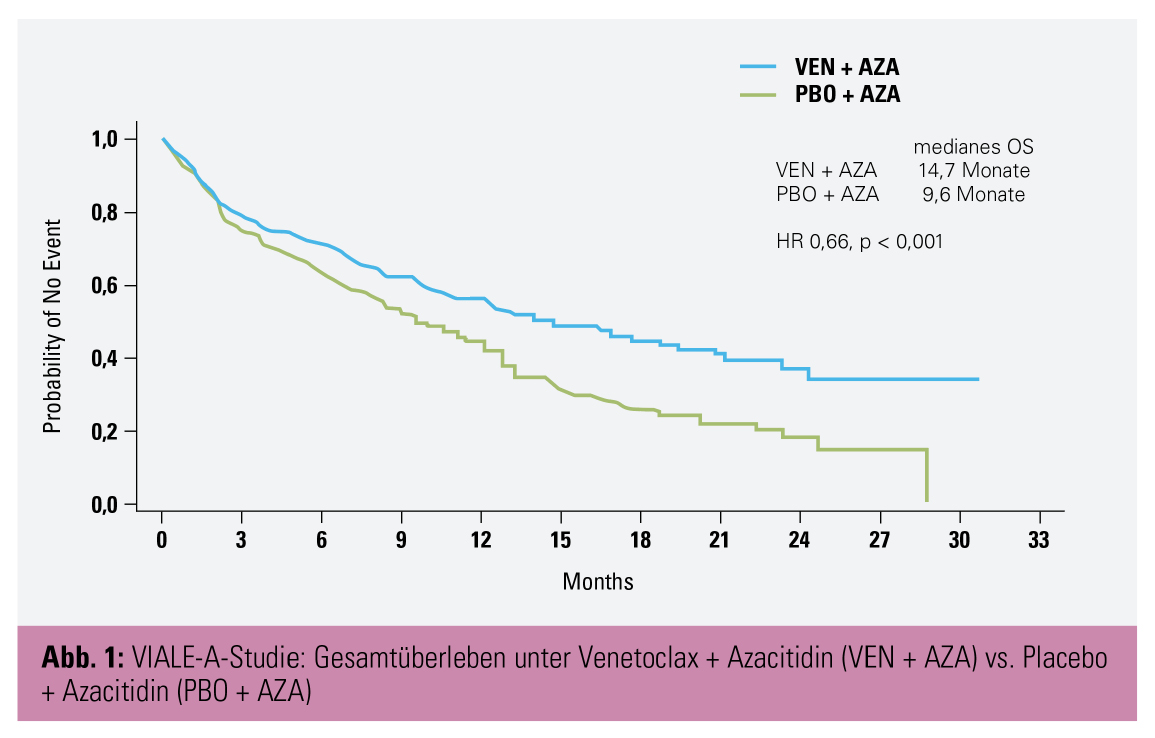
Die Kombinationstherapie war Azacitidin auch hinsichtlich des Ansprechens CR + CRi (vollständiges Ansprechen und vollständiges Ansprechen ohne Regeneration der Zellcounts) signifikant überlegen (66 vs. 28 %); das Ansprechen wurde rascher erreicht (1,2 vs. 2,8 Monate) und blieb über einen längeren Zeitraum (17,5 vs. 13,4 Monate) erhalten. Schwere unerwünschte Ereignisse umfassten febrile Neutropenie (30 vs. 10 %) und Pneumonie (16 vs. 22 %).
DiNardo C et al., A randomized, double-blind, placebo-controlled study of venetoclax with azacitidine vs. azacitidine in treatment-naive patients with acute myeloid leukemia ineligible for intensice therapy-VIALE-A. EHA 2020; Abstract LB2601
ANDROMEDA-Studie: Anti-CD38-Antikörper plus Standardtherapie bei neu diagnostizierter Leichtketten-Amyloidose
Hintergrund/klinische Rationale: Die systemische Leichtketten-(AL-)Amyloidose wird durch die Ablagerungen unlöslicher, in CD38+-Plasmazellen produzierter Amyloidfibrillen charakterisiert. Vor diesem Hintergrund untersuchte die randomisierte Open-Label-Phase-III-Studie ANDROMEDA den Effekt des Anti-CD38-Antikörpers Daratumumab (DARA) als Add-on zur Standardtherapie mit Cyclophosphamid, Bortezomib und Dexamethason (CyBorD) bei 388 Patienten mit neu diagnostizierter AL-Amyloidose mit messbarer Erkrankung, Organbeteiligung (≥ 1), kardiologischem Status I–IIIA (Mayo 2004), eGFR ≥ 20 ml/min.
Ergebnisse: Die mediane Behandlungsdauer lag bei 9,6 im DARA-CyBorD-Arm und bei 5,3 im CyBorD-Vergleichsarm. Nach einem medianen Follow-up von 11,4 Monaten wurde der primäre Endpunkt, vollständiges Ansprechen (CR), von 53 vs. 18 % der Patienten erreicht. DARA-CyBorD war dabei mit höheren Raten in Hinblick auf das hämatologische Gesamtansprechen (92 vs. 77 %) und sehr gutes partielles Ansprechen oder besser (≥ VGPR; 79 vs. 49 %). Die 6-Monats-Raten für kardiales bzw. renales Ansprechen lagen bei 42 % (vs. 22 %) bzw. 54 % (vs. 27 %). Die häufigsten Nebenwirkungen ≥ Grad 3 waren Lymphopenie, Pneumonie, Diarrhö, dekompensierte Herzinsuffizienz, Neutropenie, Synkope und periphere Ödeme.
Fazit: DARA-CyBorD erzielte bei Patienten mit neu diagnostizierter AL-Amyloidose im Vergleich mit der CyBorD-Standardtherapie höhere Responseraten, ein rascheres und tieferes hämatologisches Ansprechen und verbesserte klinische Outcomes und war mit einer akzeptablen Verträglichkeit assoziiert.
Kastritis E et al., Subcutaneous daratumumab + cyclophosphamide, bortezomib, and dexamethasone (CyBorD) in patients with newly diagnosed light chain (AL) amyloidosis: primary results from the phase 3 ANDROMEDA study. EHA 2020; Abstract LB2604
InHIBIT-Bleed: Anti-VEGF-Strategie bei hereditärer hämorrhagischer Teleangiektasie
Hintergrund/klinische Rationale: Die hereditäre hämorrhagische Teleangiektasie (HHT, Osler-Weber-Rendu-Syndrom) ist eine autosomal-dominant vererbte, seltene Erkrankung, die durch die Malformation der Blutgefäße charakterisiert ist und zu chronischen gastrointestinalen Blutungen, Epistaxis und schwerer Eisenmangelanämie führen kann. Derzeit existiert keine zugelassene Standardtherapie für diese Erkrankung. Die retrospektive Studie InHIBIT-Bleed untersuchte die Hypothese, dass der gegen VEGF gerichtete monoklonale Antikörper Bevacizumab die Ausbildung von Teleangiektasien hemmen und somit Blutungen und der resultierenden Anämie entgegenwirken kann.
Methoden: Erfasst wurden Hämoglobin, Epistaxis (anhand des Epistaxis Severity Score, ESS) und Reduktion der Erythrozyten- und Eiseninfusionen vor und nach einer Bevacizumab-Therapie (mittels T-Test bzw. Wilcoxon-Vorzeichen-Rang-Test). Die Analyse inkludierte die Daten von 238 HHT-Patienten, die in 12 internationalen Zentren im Median über 12 Monate mit Bevacizumab behandelt wurden.
Ergebnisse: Bevacizumab führte innerhalb des ersten Behandlungsjahres zu einer Verbesserung der Hämoglobin-Werte von im Mittel 3,2 g/dl und einer Reduktion des ESS um 3,4 Punkte. Verglichen mit 6 Monaten vor Therapie reduzierte Bevacizumab die Anzahl der transfundierten Erythrozyten-Einheiten um 82 %. Die Wirksamkeit der antiangiogenen Therapie war von der Schwere der Erkrankung sowie der zugrunde liegenden pathogenen Mutation (ENG vs. ACVRL1) unabhängig. Die Sicherheitsanalyse inkludierte 340,1 Patientenjahre; Bevacizumab wurde insgesamt gut vertragen, therapiebedingte Nebenwirkungen traten bei 38 % der Patienten auf (Hypertonie, Fatigue, Proteinurie, Myalgie/Arthralgie). Bei 2 % der Patienten traten venöse Thromboembolien auf, 5 % der Patienten brachen die Therapie aufgrund von Nebenwirkungen ab.
Al-Samkari H et al., An international multicenter study of systemic bevacizumab for bleeding in hereditary hemorrhagic telangiectasia: the INHIBIT-BLEED study. EHA 2020; Abstract S320
Tripeltherapie Obinutuzumab + Ibrutinib + Venetoclax im CLL-Hochrisikokollektiv
Hintergrund: Patienten mit chronisch lymphatischer Leukämie und 17p-Deletion und/oder TP53-Mutation weisen – trotz therapeutischer Fortschritte durch den Einsatz von Ibrutinib (I) und Venetoclax (Ve) – eine ungünstige Prognose auf. Obinutuzumab (G) erreichte in Kombination mit Ve in der Erstlinientherapie hohe Ansprech- und nicht nachweisbare minimale Resterkrankungs-(uMRD-)Raten, wobei del(17p)/TP53mut weiterhin negativ prognostische Faktoren darstellten.
Methoden: Die vorliegende prospektive Open-Label-Phase-II-Studie CLL2-GIVe untersuchte die Tripelkombination G + I + Ve in der Erstlinientherapie von 41 CLL-Patienten. Alle Patienten erhielten eine GIVe-Kombination über 6 Monate. Primärer Endpunkt war die CR-Rate beim finalen Re-Staging.
Ergebnisse: 26 der 41 Patienten wiesen eine del(17p) und 39 eine TP53mut auf, bei 32 Patienten lag ein negativer IgHV-Status vor. Bei 39 Patienten lag aufgrund einer hohen Tumorlast (Lymphknoten ≥ 10 cm oder ALC ≥ 25 G/l oder Splenomegalie ≥ 13 cm) und renaler Dysfunktion (CrCl < 80 ml/min) ein erhöhtes Risiko für ein Tumorlyse-Syndrom (TLS) vor. 39 Patienten erreichten das finale Re-Staging, der primäre Endpunkt wurde mit einer CR-Rate von 58,5 % erreicht (p < 0,001; Abb. 2).
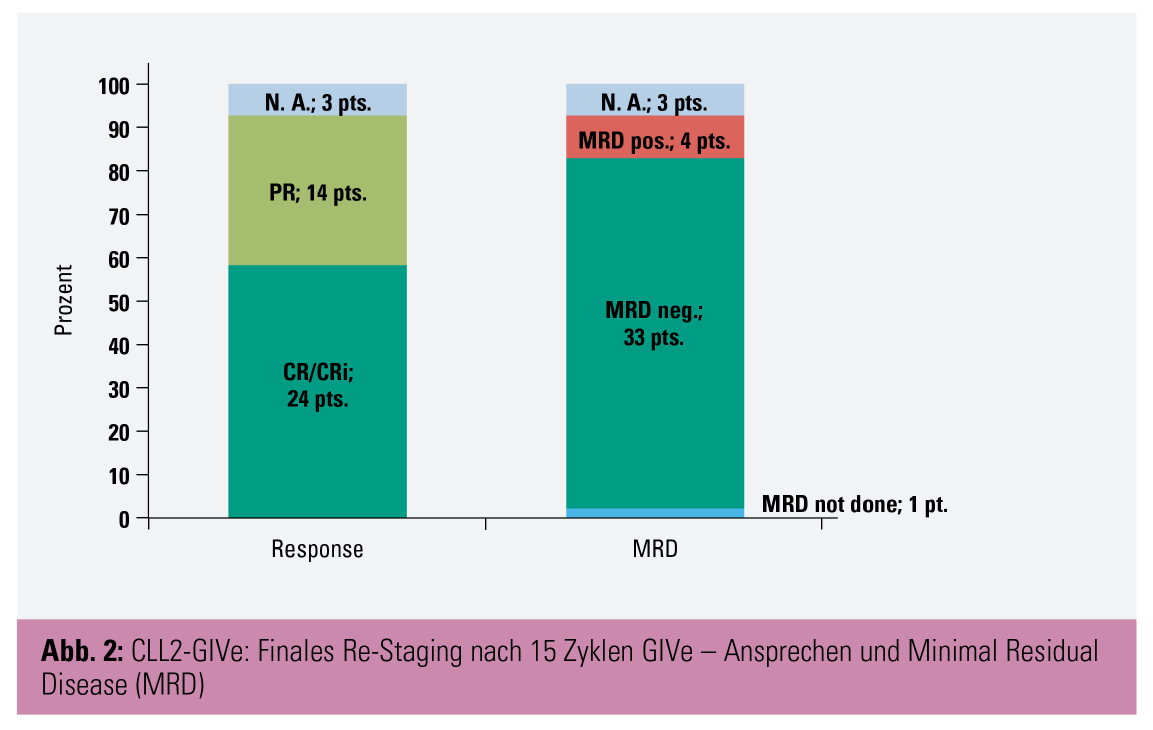
Partielles Ansprechen (PR) wurde bei weiteren 34,1 % der Patienten dokumentiert. Die häufigsten Nebenwirkungen waren gastrointestinale Beschwerden, Infektionen und Infestationen, Neutropenie und Thrombozytopenie. Kardiale Ereignisse traten bei 19,5 % der Patienten auf. Die Ergebnisse legen nahe, dass die GIVe-Tripelkombination eine vielversprechende Erstlinientherapieoption bei Patienten mit Hochrisiko-CLL darstellt.
Huber H et al., CLL2-GIVe, a prospective, open-label, multicenter phase-II trial of obinutuzumab (GA101, G), Ibrutinib (I) plus venetoclay (Ve) in untreated patients with CLL with 17p deletion/TP53 mutation. EHA 2020; Abstract S157

Ursprünglich erschienen:
UIM 06|2020
UIM 06|2020