


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Zystische Fibrose – diagnostische und therapeutische Meilensteine
28. März 2019
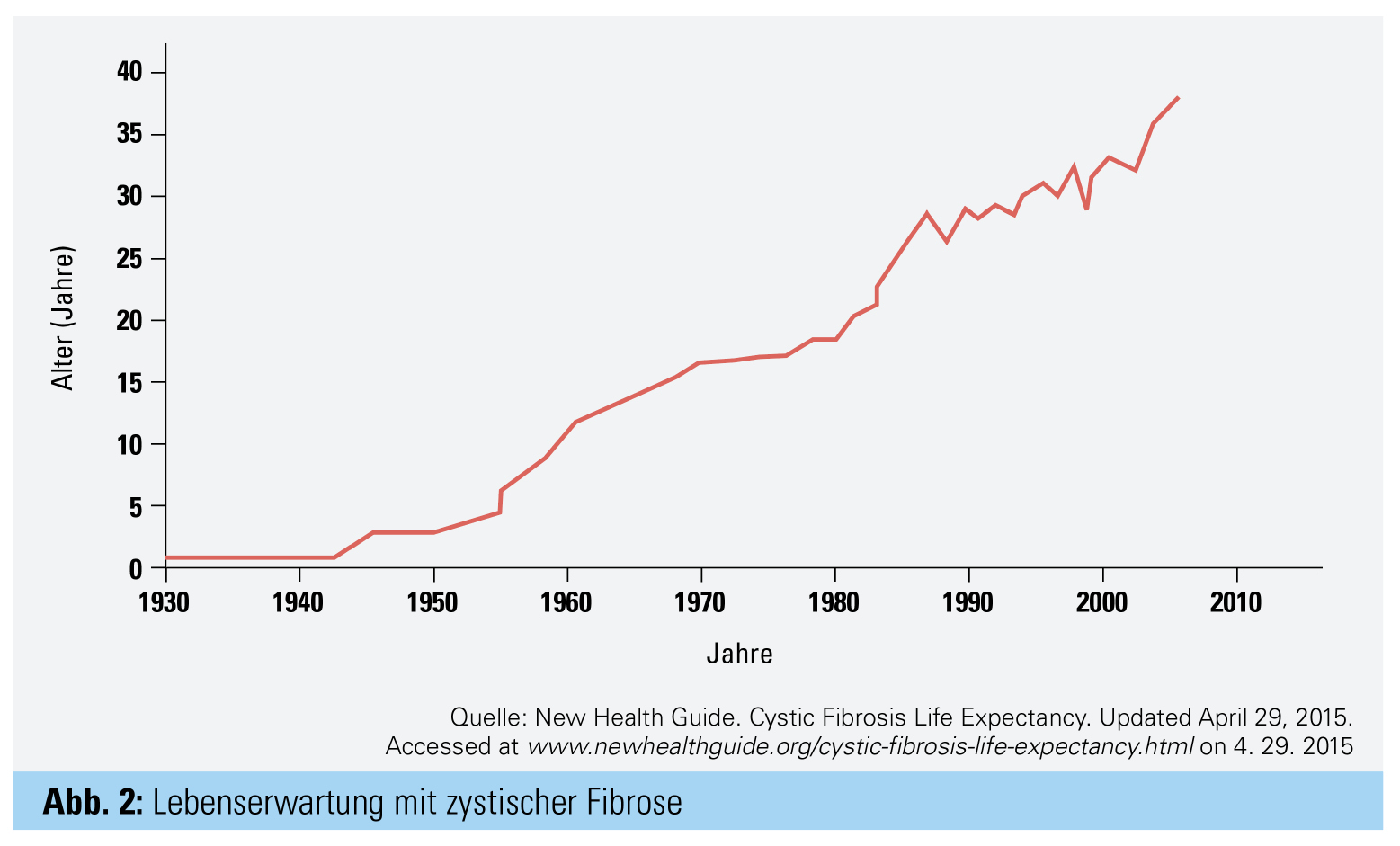
Seit der Erstbeschreibung der Erkrankung durch D. Andersen in den 1930er-Jahren ist die Lebenserwartung der an zystischer Fibrose (CF) erkrankten Kinder enorm gestiegen. Lag die Lebenserwartung in den 1950er-Jahren noch bei wenigen Monaten, so liegt diese heute im Mittel bei knapp über 40 Jahren.1 Somit ist die zystische Fibrose längst nicht mehr nur eine pädiatrische Erkrankung, sondern auch eine komplexe Multisystemerkrankung des Erwachsenenalters. Bereits heute ist annähernd die Hälfte der CF-Patienten über 18 Jahre.2
Genetik
Mit einer Prävalenz von ca. 1 : 3.000 ist die zystische Fibrose die häufigste vererbbare Stoffwechselerkrankung bei Kaukasiern. Die Vererbung ist autosomal rezessiv.
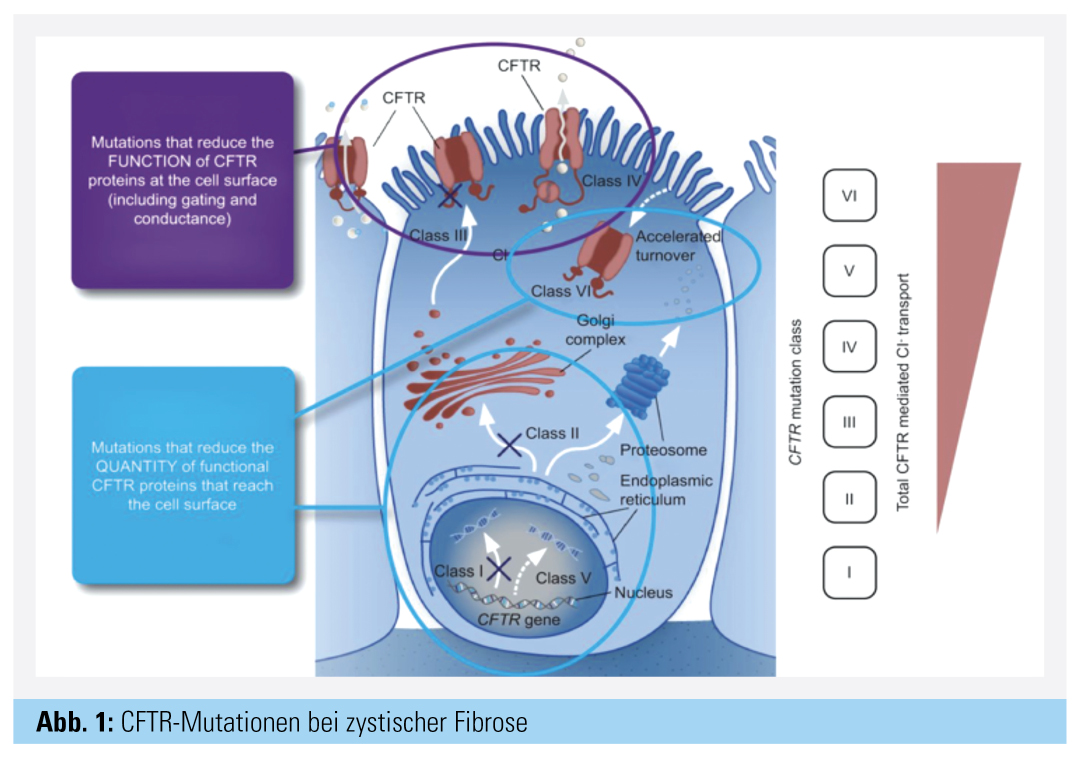
1989 gelang die Identifikation des CFTR-(Cystic Fibrosis Transmembrane Regulator-)Gens am langen Arm des Chromosoms 7. Die Mutation des Gens verursacht Funktionsstörungen des CFTR-Proteins, ein Ionenkanal auf Zellmembranen epithelialer Zellen. Mehr als 2.000 Mutationen sind derzeit bekannt, von denen etwa 300 als krankheitsauslösend gelten und in 6 Mutationsklassen eingeteilt werden.3 Die häufigste Mutation ist F508del (Klasse-II-Mutation), mit einer Häufigkeit von ca. 70 % zumindest auf einem Allel. Die Mutationsklassen I–III sind mit einem schweren Phänotyp assoziiert, während bei den Mutationsklassen IV–VI eine CFTR-Residualfunktion besteht, was eine mildere Verlaufsform bedeutet. Im Bronchialepithel reduziert der CFTR-Defekt die intraluminale Sekretion von Chloridionen. Dies bedingt die Dehydratation der mukoziliären Schicht mit eingeschränkter mukoziliärer Clearance. Folglich kommt es zu zähem Sekret, bakterieller Besiedelung und chronischer Inflammation mit progredienter Ausbildung von Bronchiektasien. Neben der Lunge sind auch Pankreas (endokrine und exokrine Pankreasinsuffizienz), Leber (Hepatopathie), Verdauungstrakt (Malabsorption, Ileus), Vas deferens (Infertilität) und Nasennebenhöhlen (chronische Sinusitis) betroffen.
Diagnostik
Ein diagnostischer Meilenstein war die Etablierung eines Neugeborenscreenings (in Österreich seit 1998 verfügbar). Bei diesem Test handelt es sich um einen zweistufigen Test, wobei immunreaktives Trypsinogen (IRT) im Blut des Neugeborenen bestimmt wird. Ist IRT auch im zweiten Test erhöht, wird die Diagnose mittels Schweißtest bestätigt.
Nach einem positiven oder intermediärem Testergebnis im Schweißtest muss eine molekulargenetische Untersuchung zum Nachweis der zugrunde liegenden CFTR-Mutation erfolgen.4
Dennoch verläuft in Österreich bei ca. einem Kind pro Jahr das Screening falsch negativ, und die Diagnose erfolgt verzögert, wobei klinische Symptome in diesem Fall hinweisend sind (fettreiche Stühle, Gedeihstörung, rezidivierende Bronchitiden und Pneumonien). Die frühe Diagnosestellung erlaubt den frühen Beginn einer multidisziplinären Betreuung in einem CF-Zentrum.
Therapie
Die Säulen der Therapie sind Atemphysiotherapie, inhalative Therapien, Ernährungstherapie, Antibiotika sowie als modernster kausaler Therapieansatz sogenannte CFTR-Modulatoren. Der erste therapeutische Meilenstein in der CF-Behandlung war mit der Substitution von Pankreasenzymen in den 1950er-Jahren gelegt, denn eine Verbesserung des Ernährungsstatus ist mit einer deutlich besseren Prognose assoziiert.1, 5
Ein weiterer Durchbruch gelang mit der Verfügbarkeit der inhalativen Antibiotika Tobramycin und Colistin, die einerseits zur Suppressionstherapie bei chronischer Besiedelung mit Pseudomonas aeruginosa dienen und in der Eradikationstherapie bei der Erstinfektion einen bedeutenden Stellenwert einnehmen. Eine chronische Pseudomonas-aeruginosa-Infektion ist mit einem rascherem Lungenfunktionsabfall, schlechterem Ernährungsstatus und häufigeren Exazerbationen assoziiert.
Heute stehen neben Colistin und Tobramycin auch Aztreonam-Lysin und Levofloxacin zur inhalativen Suppressionstherapie bei chronischer Pseudomonas-Besiedelung zur Verfügung6, die in unterschiedlichen Schemen miteinander kombiniert werden können.
Ein fixer und wesentlicher Bestandteil der Behandlung ist die Inhalationstherapie (hypertone Kochsalzlösungen, DNAse, Bronchodilatatoren) und Atemphysiotherapie zur Mobilisierung von zähen Sekretmassen. Während früher Klopfmassagen üblich waren, umfasst die moderne Atemphysiotherapie differenzierte und personalisierte Techniken wie autogene Drainage, Flutter- und PEP-Therapie. Unerlässlich ist auch ein regelmäßiges Ausdauer- und Krafttraining sowie in fortgeschrittenem Stadium Atemmuskeltraining. Somit liegt es auf der Hand, dass der Therapiealltag eines CF-Patienten enorm zeit- und energieaufwendig ist und immer wieder viel Motivation durch das Behandlungsteam braucht.
Ein immer größeres Problem stellen Infektionen mit multiresistenten gramnegativen Keimen, MRSA, Burkholderia-cepacia-Komplex und atypischen Mykobakterien (M. abscessus) dar – nicht nur therapeutisch durch die begrenzte Verfügbarkeit von Antibiotika, sondern auch im Hygienemanagement mit der Notwendigkeit einer strengen Separation in den Ambulanzen und stationären Bereichen. Die Isolation kann beim einzelnen Patienten zu einer enormen psychosozialen Belastung führen.
CFTR-Modulatoren: Mit der Entwicklung von CFTR-Modulatoren gelang ein weiterer Meilenstein in Richtung kausaler Therapie. 2012 wurde als erster CFTR-Modulator Ivacaftor bei einigen Klasse-III-Mutationen (sog. Gating-Mutationen) zugelassen. Ivacaftor ist ein „Potenziator“ und erhöht die Kanalöffnungswahrscheinlichkeit für Cl-Ionen an der Zellmembran.7 Lumacaftor als „Korrektor“ verbessert hingegen die Prozessierung und den Transport von CFTR-Molekülen und erhöht somit die Menge an funktionstüchtigen Ionenkanälen an der Zelloberfläche. Mit dem Kombinationspräparat eines „Korrektors“ und eines „Potenziators“ (Ivacaftor/Lumacaftor) stand wenig später für F508del-homozygote Patienten eine weitere kausale Behandlungsoption zur Verfügung. Mit CFTR-Modulatoren zeigte sich eine Verbesserung der FEV1, Reduktion von Exazerbationen und i. v. Therapien sowie deutliche Symptomverbesserung im Sinne von weniger Husten und geringeren Sputummengen.8, 9 Eine Langzeitbeobachtungsstudie über 92 Wochen ergab einen moderaten, statistisch nichtsignifikanten Effekt hinsichtlich Reduktion von Exazerbationsraten und Hospitalisierungen.10
Kürzlich wurde mit Tezacaftor/Ivacaftor eine weitere CFTR-Modulatoren-Kombination für homozygote F508del sowie heterozygote F508del in Kombination mit Mutationen mit einer Restfunktion (bei Patienten > 12 Jahre) zugelassen. Tezacaftor/Ivacaftor scheint sich einerseits durch eine deutlichere Verbesserung der FEV1 gegenüber dem Ausgangswert, ein besseres Nebenwirkungsprofil hinsichtlich Erhöhung von Leberfunktionsparametern sowie respiratorischen Nebenwirkungen und weniger Interaktionen mit anderen Medikamenten auszuzeichnen.11, 12
Trotz aller Fortschritte ist die progrediente respiratorische Insuffizienz die häufigste Todesursache bei CF-Patienten. Wenn alle konservativen Therapiemaßnahmen ausgeschöpft sind, sollte eine Lungentransplantation (LuTX) in Erwägung gezogen werden. Auch das Outcome nach einer LuTX hat sich in den letzten Jahrzehnten drastisch verbessert. So liegt die perioperative Letalität durch Verbesserung der chirurgischen und intensivmedizinischen Methoden sowie der immunsuppressiven Therapie bei 10–15 %. Die 5-Jahres-Überlebensrate liegt international bei 55 %, wobei große Zentren im Langzeitverlauf wesentlich bessere Ergebnisse erzielen, was auf die unterschiedliche Effizienz in der Nachsorge schließen lässt.13
Resümee
Durch bedeutende diagnostische und therapeutische Fortschritte hat die mediane Lebenserwartung von CF-Patienten enorm zugenommen. Die Erkrankung ist längst in der Erwachsenen-Pneumologie angekommen und bedarf einer standardisierten, multidisziplinären Betreuung in einem spezialisierten CF-Zentrum.
1 Elborn JS, Cystic fibrosis. Lancet 2016; 388:2519–2531
2 Burgel PR et al., Future Trends in Cystic Fibrosis Demography in 34 European Countries. Eur Respir J 2015; 46:133–141
3 http://www.CFTR2.org
4 S2-Konsensus-Leitlinie „Diagnose der Mukoviszidose“ (AWMF 026-023) unter Federführung der Gesellschaft für Pädiatrische Pneumologie
5 Kerem E et al., Factors associated with FEV1 decline in Cystic Fibrosis: Analysis of the ECFS patient registry. Eur Respir J 2014; 43(1):125–133
6 Castellani et al., ECFS Best Practice Guidelines: The 2018 Revision. J Cyst Fibros 2018; 17:153–178
7 Ramsey BW et al., A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N Engl J Med 2011; 365:1663–1672
8 Wainwright CE et al., Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl Med J 2015; 373:220
9 McColley SA et al., Lumacaftor/Ivacaftor reduces pulmonary Exacerbations in Patients irrespective of initial Changes in FEV1. J Cyst Fibros 2019; 18:94–101
10 Konstan MW et al., Assessment of Safety and Efficacy of long-term Treatment with Combination Lumacaftor and Ivacaftor Therapy in Patients with Cystic Fibrosis Homozygous for the F508del-CFTR Mutation (PROGRESS): a Phase 3, Extension study. Lancet Respir Med 2017; 5:107–118
11 Taylor-Cousar JL et al., Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017; 377:2013–2023
12 Rowe SM et al., Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med 2017;377:2024–2035
13 Gottlieb J et al., Lungentransplantation bei Mukoviszidose – ein Positionspapier Pneumologie 2009; 63:451–460

AutorIn: Dr. Andrea Lakatos-Krepcik
CF-Zentrum für Erwachsene, Abteilung für Atmungs- und Lungenerkrankungen, Krankenhaus Hietzing, Wien

Ursprünglich erschienen:
UIM 02|2019
UIM 02|2019
