
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Arginase-1-Defizienz
Bei Spastik an Harnstoffzyklusdefekt denken
31. Oktober 2025
Arginase-1-Defizienz ist eine seltene, autosomal-rezessiv vererbte Harnstoffzyklusstörung, bei der das Enzym Arginase 1 durch eine Mutation im Arginase-1-Gen nicht oder nur unzureichend gebildet wird. Dadurch kommt es zu einer Anreicherung von Arginin und seiner Stoffwechselprodukte mit weitreichenden metabolischen Folgen, die vor allem das zentrale Nervensystem betreffen.
Erstmanifestationen, Krankheitsverlauf und Red Flags
Die Symptome von Kindern und Jugendlichen mit Arginase-1-Mangel haben zwei Häufigkeitsgipfel. Der erste findet sich im Neugeborenenalter (Abb.), wobei die Symptome hier auf eine Erhöhung von Ammoniak zurückzuführen sind. Weil Arginase fehlt, staut sich Ammoniak als Abbauprodukt an, das toxisch ist, insbesondere für das Gehirn. Das erste Symptom bei Neugeborenen ist häufig ein hyperammonämisches Koma, beginnend mit Bewusstseinsstörungen bis hin zu einer Multisystemerkrankung. Diese hyperammonämischen Krisen können auch später auftreten, sind aber mit zunehmendem Alter der Kinder seltener. Das hat 2 Gründe: erstens, weil die Diagnose dann idealerweise bereits bekannt ist, und zweitens, weil man die Stoffwechselstörung therapiert.
Der zweite Häufigkeitsgipfel ist auf die Auswirkungen der erhöhten Argininkonzentration zurückzuführen. Hier geht es um die anderen Stoffwechselwege im Körper, bei denen Arginin beteiligt ist. Das äußert sich in Form einer fortschreitenden Zerebralparese:
Die Kinder zeigen etwa im Kindergartenalter eine Spastik – zuerst mit erhöhtem Tonus der unteren Extremitäten. Später können auch die oberen Extremitäten betroffen sein. Sie haben zunehmende Geh- und Koordinationsprobleme, bis sie die Gehfähigkeit verlieren und auf einen Rollstuhl angewiesen sind (Abb.).
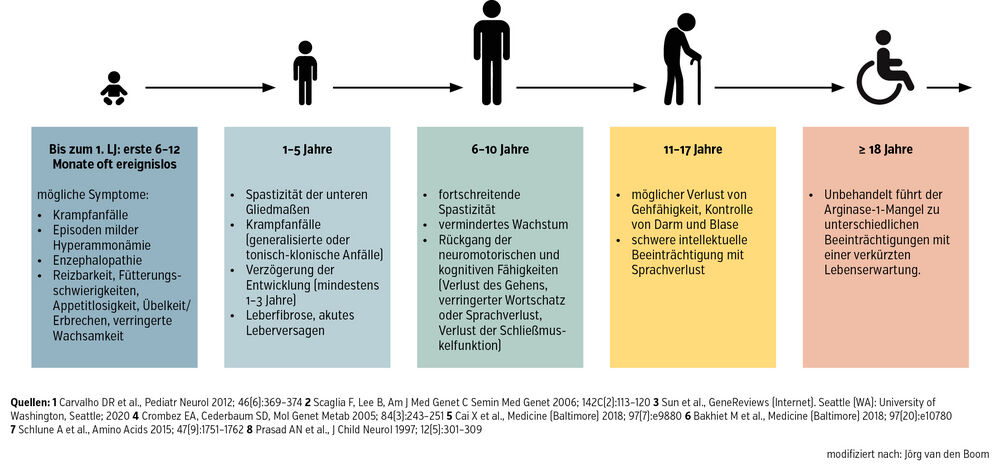
Abb.: Verlauf und Symptome bei Arginase-1-Defizienz
Es gibt auch Kinder, die nur die neurologische Beteiligung zeigen, ohne dass sie jemals eine hyperammonämische Krise hatten. Wenn diese Betroffenen im Neugeborenenalter keine Symptome zeigen, jedoch im Laufe ihres Lebens eine neurologische Symptomatik entwickeln, kann es sein, dass sie länger oder gänzlich als Arginase-1-Mangel-Patient:innen unentdeckt bleiben.
Daher: Wenn ein Kind eine progrediente Spastik zeigt, sollte immer die Bestimmung von Arginin im Plasma erfolgen! Weil die Symptome sehr unspezifisch sein können, es aber eine Therapiemöglichkeit gibt, ist der Arginase-1-Mangel in einigen Ländern bereits seit etlichen Jahren im Neugeborenen-Screening inkludiert. In Österreich wird seit 2018 auf diese angeborene Stoffwechselstörung gescreent.
Differenzialdiagnostische Abgrenzung
Differenzialdiagnostisch abzugrenzen sind insbesondere andere Harnstoffzyklusdefekte, etwa Defizite der Ornithintranscarbamylase, Argininosuccinatlyase oder Citrullinämie Typ I, die jedoch meist neonatale hyperammonämische Krisen und andere Aminosäuremuster zeigen (Arginin in der Regel erniedrigt). Da die klinische Symptomatik oft jener einer zerebralen Bewegungsstörung ähnelt, kommen auch hereditäre spastische Paraplegien oder Leukodystrophien infrage. Die Bestimmung des Aminosäureprofils im Plasma mit Nachweis stark erhöhter Argininkonzentration ermöglicht eine klare biochemische Abgrenzung.
Diagnosepfad
Wichtig ist, dass bei der Symptomkonstellation (Abb.) an das mögliche Vorliegen einer angeborenen Harnstoffzyklusstörung gedacht wird. Bei einem entsprechenden Verdacht erfolgt die Sicherung der Diagnose durch die Bestimmung der Aminosäuren im Plasma. Bei Betroffenen ist die Argininkonzentration deutlich erhöht: Der Referenzwert liegt bei bis zu 120 Mikromol pro Liter (µmol/l) im Blut, bei Patient:innen mit Arginase-1-Mangel kann der Wert auch einmal bei 500 µmol/l bis über 1.000µmol/l liegen. Die Ergebnisse können anschließend mit einer genetischen Analyse gesichert werden, indem man das Arginase-1-Gen analysiert.
Therapiemöglichkeiten
Die Standardbehandlung besteht in einer eiweißdefinierten Ernährung, bei der man darauf achtet, dass die Argininzufuhr nicht durch die Ernährung zusätzlich erhöht wird. Sie stellt eine strenge Form der Diät dar, bei der Protein deutlich eingeschränkt werden muss. Zudem kann man sogenannte Ammoniak-Scavenger p. o. verabreichen (Natriumbenzoat, Glycerolphenylbutyrat). Sie „fangen“ Ammoniak aus dem Blutkreislauf ab und entgiften dadurch das Blut. Seit 5 Jahren steht für den Arginase-1-Mangel eine Enzymtherapie zur Verfügung: Durch wöchentliche subkutane Spritzen des Medikamentes Pegzilarginase wird das Arginin im Blut abgebaut, zu Ornithin umgewandelt und der Argininspiegel unter 200 µmol/l gesenkt. Die Pegzilarginase ersetzt allerdings nicht die Aktivität der Arginase in der Leber, so dass eventuell weiterhin mit Einschränkungen, die den Harnstoffzyklus betreffen, gerechnet werden muss.
Die zuvor genannte eiweißarme Diät ist eine sehr einschränkende Diät, welche die Symptome zwar verlangsamt, aber nicht verhindert. Daher werden große Hoffnungen auf die Enzymtherapie gesetzt, weil darunter die Eiweißzufuhr in der Nahrung erhöht werden kann. Es ist zu erwarten, dass die Pegzilarginase-Therapie auch im Langzeitverlauf die neurologischen Symptome abbremsen und im besten Falle sogar verhindern kann. Bei frühzeitigem Beginn würden potenzielle Folgeschäden verhindert, was für Betroffene ein Plus an Lebensqualität bedeuten würde.
Bei allen seltenen Erkrankungen lautet das Motto:
„Nicht der/die Patient:in soll reisen, sondern die Expertise.“
Die generelle Patientenbetreuung sollte so gut und wohnortnah wie möglich erfolgen. Das ist übrigens auch ein Vorteil der subkutanen wöchentlichen Enzymtherapie, denn diese kann zuhause und selbstständig verabreicht werden.

Autorin:
Ao. Univ.-Prof.in Dr.in Daniela Karall
Ao. Univ.-Prof.in Dr.in Daniela Karall
Präsidentin der ÖGKJ

Autor:
PDin Dr.in med. Dipl-oec-troph.in Sabine Scholl-Bürgi
PDin Dr.in med. Dipl-oec-troph.in Sabine Scholl-Bürgi
Zweite Sekretärin der Österreichischen Gesellschaft für Kinder- und Jugendheilkunde

Ursprünglich erschienen:
AEK SH Seltene Erkrankungen|2025
AEK SH Seltene Erkrankungen|2025
KRANKHEITSSTECKBRIEF
- Der Arginase-1-Mangel ist von den 6 bekannten Harnstoffzyklusdefekten der seltenste und tritt mit einer Häufigkeit von nur einer betroffenen Person unter 750.000 bis 900.000 Menschen auf.
- Betroffene weisen eine Mutation im Arginase-1-Gen auf, was dazu führt, dass das Enzym Arginase 1 in zu geringen Mengen oder gar nicht produziert wird. Dieses Enzym wird benötigt, um Arginin und argininverwandte Stoffwechselprodukte abzubauen.
- Die Besonderheit des Arginase-1-Mangels ist, dass Arginin nicht nur Teil des Harnstoffzyklus, sondern z. B. auch des Energiestoffwechsels ist und bei der Bildung von Kreatin und Kreatinphosphat eine Rolle spielt sowie am NO-Synthese-Weg und in der Polyaminbildung beteiligt ist.
- Klinisch reichen die Symptome von Krampfanfällen, Spastizität und neurologischen Problemen bis zu vermindertem Wachstum und Entwicklungsverzögerungen. Im weiteren Verlauf kommt es zum Verlust der Gehfähigkeit und schweren kognitiven Beeinträchtigungen; unbehandelt ist mit einer deutlich verkürzten Lebenserwartung zu rechnen.
- Die Behandlung umfasst eine proteinarme Diät, die Verabreichung von Stickstofffängern sowie eine Therapie mit Pegzilarginase, wodurch das Arginin im Blut abgebaut werden kann.
Literatur bei den Verfasserinnen