






















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Kardiale Transthyretin-Amyloidose
Im klinischen Alltag angekommen
31. Oktober 2025
Die kardiale Transthyretin-Amyloidose (ATTR) ist eine infiltrative Kardiomyopathie, die entweder genetisch bedingt (ATTRv) oder altersassoziiert (ATTRwt) auftritt.
Erstmanifestationen, Red Flags und Verdachtsmomente bei ATTR
Die Symptomatik der ATTR ist heterogen und hängt wesentlich von den jeweils betroffenen Organen sowie dem Ausmaß der Ablagerungen ab. Eine kardiale Beteiligung äußert sich häufig unspezifisch in Form einer Herzinsuffizienz mit Belastungsdyspnoe, Müdigkeit und peripheren Stauungszeichen. Auch Rhythmusstörungen – bradykard oder tachykard – können auftreten und sind potenziell mit Schwindel, Synkopen oder sogar plötzlichem Herztod verbunden.
Wichtig ist zudem, dass extrakardiale Manifestationen einer späteren Herzbeteiligung oft zeitlich deutlich vorausgehen. Insbesondere Beschwerden des muskuloskelettalen Systems oder ein ein-/beidseitiges Karpaltunnelsyndrom gelten als charakteristische Frühzeichen und sollten den Verdacht auf eine ATTR, insbesondere im Beisein anderer Red Flags, erhärten (Tab.).
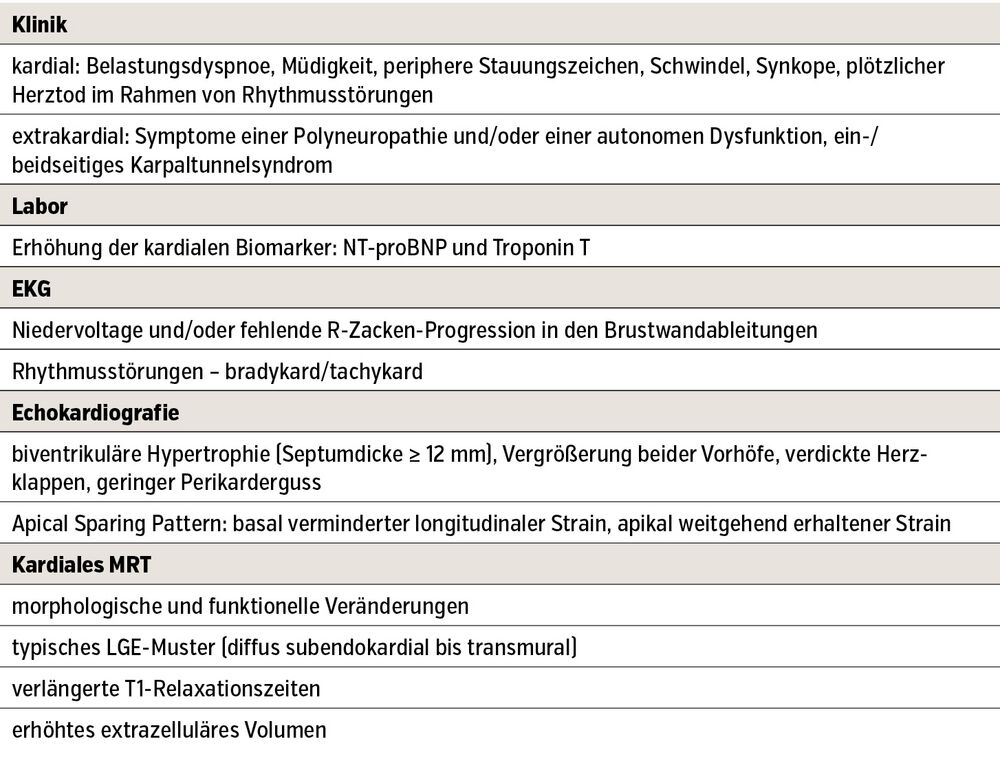
Tab.: Red Flags für die kardiale ATTR-Amyloidose
Diagnostikpfad bei ATTR
Echokardiografie: Besteht klinisch der Verdacht auf eine Amyloidose, liefert die Echokardiografie in vielen Fällen erste richtungsweisende Befunde. In frühen Krankheitsstadien ist meist eine diastolische Funktionsstörung nachweisbar, während im Verlauf zusätzlich die systolische Funktion beeinträchtigt ist. Typische echokardiografische Merkmale umfassen eine biventrikuläre Hypertrophie, eine Vergrößerung beider Vorhöfe, verdickte Herzklappen sowie hämodynamisch nicht wirksame Perikardergüsse. Ein charakteristisches Zeichen stellt das „apical sparing pattern“ dar, bei dem der longitudinale Strain basal vermindert, apikal jedoch weitgehend erhalten ist.
Die kardiale Magnetresonanztomografie (MRT) ist zur Differenzierung gegenüber anderen Herzmuskelerkrankungen bedeutsam. Sie zeigt morphologische und funktionelle Veränderungen, ein typisches LGE-Muster (Late Gadolinium Enhancement), verlängerte T1-Zeiten und ein erhöhtes extrazelluläres Volumen (Tab.). Weder Echokardiografie noch MRT ermöglichen jedoch eine definitive Diagnosestellung.
Durch die Kombination aus negativer Leichtkettendiagnostik und positiver Skelettszintigrafie ist heute meist dennoch eine sichere Diagnose nichtinvasiv möglich. Erstere (freie Leichtketten, Serum-/Harneiweißelektrophorese mit Immunfixation) schließt eine AL-Amyloidose aus, die unbehandelt prognostisch äußerst ungünstig ist. Bei der Skelettszintigrafie werden radioaktive Tracer (DPD, PYP, HMDP) eingesetzt. Die Stärke des myokardialen Tracer-Uptakes wird nach Perugini (0–3) bewertet. Eine deutliche Tracer-Anreicherung (Perugini ≥ 2) mit einer negativen Leichtkettendiagnostik sichert die ATTR-Diagnose. Anschließend wird unabhängig vom Patientenalter eine genetische Untersuchung empfohlen.
Wann Biopsie? Sind die Kriterien einer nichtinvasiven Diagnosestellung nicht erfüllt (positive Leichtketten und/oder Perugini <2), ist bei fortbestehendem klinischem Verdacht eine Biopsie erforderlich. Das gilt ebenso für die seltene, aber nicht auszuschließende Koinzidenz von positiven Leichtketten und Perugini ≥ 2. Je nach Zentrumserfahrung erfolgt eine Endomyokard- oder extrakardiale Biopsie mit Amyloidtypisierung.
Therapie der ATTR
Die symptomatische Behandlung fokussiert auf die Kontrolle der Herzinsuffizienz. Basis sind Schleifendiuretika mit dem Ziel einer Euvolämie. Ergänzend können Aldosteronantagonisten und SGLT2-Inhibitoren eingesetzt werden. RAAS-Blocker und Beta-Blocker sind wegen Hypotonie und chronotroper Inkompetenz oft schlecht verträglich. Aufgrund des hohen Schlaganfallrisikos wird bei Vorhofflimmern unabhängig vom CHA2DS2-VA-Score eine orale Antikoagulation empfohlen.
Krankheitsspezifische Therapien greifen in die Amyloid-Pathogenese ein. TTR-Stabilisatoren (Tafamidis, Acoramidis) verhindern die Dissoziation der Tetramere. Gene Silencer wie Patisiran, Vutrisiran, Inotersen und Eplontersen sind für ATTRv mit Polyneuropathie etabliert. Für Vutrisiran konnte die Wirksamkeit auch bei kardialer Manifestation (ATTRwt und ATTRv) gezeigt werden.
Innovative Therapieansätze werden darüber hinaus derzeit in klinischen Studien untersucht, wie ein monoklonaler Antikörper (NI006) sowie ein CRISPR/Cas9-basiertes Verfahren (Nexiguran-Ziclumeran, Nex-Z). Besonders die Antikörpertherapie gilt als vielversprechend, da diese durch gezielte Amyloid-Clearance potenziell eine strukturelle und funktionelle Verbesserung betroffener Organe ermöglichen könnte und damit ein kurativer Therapieansatz ist.

Autorin:
Dr.in Maria Ungericht, PhD
Dr.in Maria Ungericht, PhD
Universitätsklinik für Innere Medizin III – Kardiologie und Angiologie, Medizinische Universität Innsbruck

Autor:
Univ.-Prof. Dr. Gerhard Pölzl, FESC, FHFA
Univ.-Prof. Dr. Gerhard Pölzl, FESC, FHFA
Universtitätsklinik für Innere Medizin III
Kardiologie und Angiologie, Interdisziplinäres Herzinsuffizienzzentrum Tirol – IHZ, Zentrum für seltene Herzmuskelerkrankungen, Medizinische Universität Innsbruck

Ursprünglich erschienen:
AEK SH Seltene Erkrankungen|2025
AEK SH Seltene Erkrankungen|2025
Krankheitssteckbrief: kardiale Transthyretin-Amyloidose
- Ursache der ATTR ist die Instabilität des in der Leber produzierten Transthyretins (TTR). TTR-Tetramere zerfallen in der Folge in Monomere, die zu unlöslichen Fibrillen aggregieren und sich im Interstitium des Myokardes ablagern.
- Die Folge sind Verdickung und Steifigkeit des Herzmuskels. Auch extrakardiale Ablagerungen sind möglich.
- Die Erkrankung blieb in der Vergangenheit häufig unerkannt und war mit einer hohen Sterblichkeit verbunden.
- In den letzten Jahren haben neue diagnostische Verfahren und therapeutische Entwicklungen dazu beigetragen, dass die ATTR heutzutage früher erkannt wird und besser behandelt werden kann.
- Differenzialdiagnose AL-Amyloidose: Ausschluss durch negative Leichtkettendiagnostik.
WISSENSWERTES FÜR DIE PRAXIS
- Jede unklare Myokardverdickung ≥ 12 mm bedarf einer weiteren Abklärung auf Amyloidose, insbesondere wenn zusätzliche Red Flags vorliegen (Tab.).
- Diagnose mittels DPD-Szintigrafie, Ausschluss freie Leichtketten/Serum-/Harneiweißelektrophorese mit Immunfixation, evtl. kardiales MRT.
- Zuweisung an spezialisierte Zentren zur weiteren Abklärung und Therapie bei begründetem Verdacht.
Literatur bei den Verfasser:innen
Bildnachweis
Vorschaubild: © Petro – stock.adobe.com