


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hypertrophie im Ultraschall und Karpaltunnelsyndrom in der Anamnese
Kardiale Transthyretin-Amyloidose
22. September 2023
Die kardiale ATTR-Amyloidose (Amyloidose auf Basis des Transportproteins Transthyretin) schien lange eine seltene Erkrankung zu sein.
Im Jahr 2016 wurde ein Diagnosealgorithmus (Gilmore-Algorithmus) etabliert, der bei Patient:innen mit typischen Symptomen, echokardiografischen Veränderungen, positivem DPD-Knochenscan und laborchemischem Ausschluss einer Leichtketten-Amyloidose eine Diagnosestellung ohne Biopsie erlaubt. Seither ist klar, dass die Erkrankung relativ häufig ist, beispielsweise beträgt die Prävalenz unter allen Patient:innen, die zu einer perkutanen Aortenklappenimplantation (TAVI) zugewiesen werden, 14 %, unter Patient:innen, die als hypertrophe Kardiomyopathie laufen, 6 %.
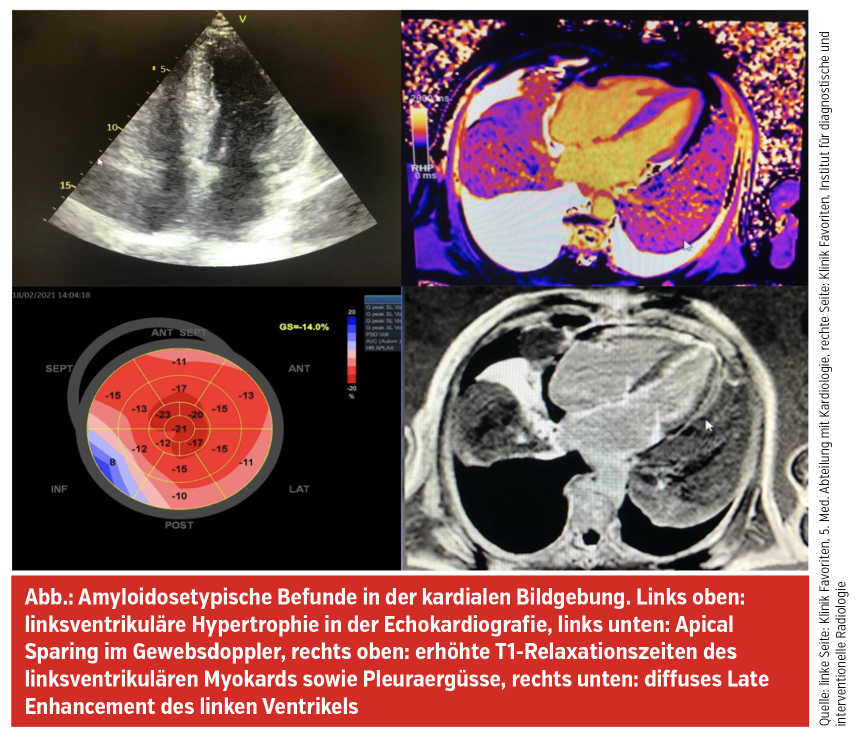
Hinweise, die an eine kardiale ATTR-Amyloidose denken lassen sollen, sind insbesondere eine links- und rechtsventrikuläre Hypertrophie in der Echokardiografie bei fehlendem Hypertrophiezeichen im EKG und die Anamnese eines (oft vor > 10 Jahren operierten) Karpaltunnelsyndroms. Im EKG finden sich häufig auch Pseudoinfarktmuster, Vorhofflimmern, AV-Blockierungen (bzw. dann ein Z. n. Schrittmacherimplantation) oder periphere Niedervoltage durch die interstitiellen TTR-Amyloidfibrillen. Echokardiografisch erhärten diastolische Funktionsstörungen, ein kleiner Perikarderguss und insbesondere auch das Apical Sparing (erhaltener Strain im Apex bei reduzierten Werten in den mittleren und basalen Segmenten) den Verdacht auf eine ATTR-Amyloidose. Von dieser Erkrankung abzugrenzen ist die AL-Amyloidose. Dabei handelt es sich um eine hämatoonkologische Erkrankung, bei der es in Plasmazellen zur Produktion von monoklonalen Leichtketten kommt, die sich in weiterer Folge systemisch ablagern, wobei Niere und Herz besonders betroffen sind. Die Patient:innen fallen daher mit Proteinurie, linksventrikulärer Hypertrophie oder NT-pro-BNP-Anstieg auf.
Im Rahmen der weiteren Abklärung und Differenzierung erfolgt ein DPD-Knochenscan, der myokardiale TTR-Ablagerungen bestätigen kann, eine laborchemische Untersuchung hinsichtlich freier Leichtketten in Harn und Serum sowie eine kardiale Magnetresonanztomografie, in der neben den oben genannten morphologischen kardialen Veränderungen deutlich erhöhte T1-Relaxationszeiten und ein typisches diffuses linksventrikuläres Late Enhancement nachgewiesen werden kann. Mit Hilfe dieser Untersuchungen kann eine ATTR-Amyloidose in aller Regel nichtinvasiv diagnostiziert werden, Myokardbiopsien sind heutzutage nur selten notwendig. Schließlich erfolgt eine Untersuchung hinsichtlich Mutationen im TTR-Gen, da für die seltene hereditäre ATTRv-Amyloidose erweiterte Behandlungsmöglichkeiten im Vergleich zur Wildtyp-Form (ATTRwt-Amyloidose) zur Verfügung stehen und bei dieser häufiger rasch progrediente Polyneuropathien auftreten.
Therapieoptionen
Die führende spezifische Therapie der ATTR-Amyloidose (Wildtyp und hereditäre Form) ist die Stabilisierung des Transthyretintetramers mittels Tafamidis. Damit kann die weitere ATTR-Ablagerung stark verzögert werden. Bei der hereditären Form der Amyloidose ist ab Vorhandensein einer Polyneuropathie im Stadium 1 oder 2 alternativ auch eine Gene-Silencer-Therapie mittels Patisiran (i. v.), Vutrisiran (s. c.) oder Inotersen (s. c.) nach Verordnung durch Fachärzt:innen für Neurologie indiziert. In Zukunft werden diese Therapien auch für die Wildtyp-ATTR-Amyloidose zugelassen werden. An neuen Behandlungsansätzen, um auch bereits abgelagertes Transthyretin-Amyloid z. B. mittels Chaperonen oder Antikörpern aus den Geweben zu entfernen, wird derzeit geforscht.

Autorin:
Assoz. Prof.in Priv.-Doz.in Dr.in Diana Bonderman
Assoz. Prof.in Priv.-Doz.in Dr.in Diana Bonderman
5. Medizinische Abteilung mit Kardiologie und internistische Notaufnahme, Klinik Favoriten, Wien

Autorin:
DDr.in Silvia Charwat-Resl
DDr.in Silvia Charwat-Resl
5. Medizinische Abteilung mit Kardiologie, Klinik Favoriten, Wien

Ursprünglich erschienen:
AEK 18|2023
AEK 18|2023
Was Patient:innen wissen möchten
Bei mir lagert sich ein fehlgefaltetes Protein im Herzen ab, kann ich mit Ernährung die Ablagerungen stoppen?
Nachdem die Transthyretin-Amyloidose sowohl das periphere als auch das autonome Nervensystem befällt und so im Krankheitsverlauf zu vermindertem Appetit/Völlegefühl führt, ist eine ausgewogene Ernährung wichtig. Es gibt aber keine verbotenen Speisen. Für Grünteepräparate gibt es keine überzeugende Evidenz, dass sie die Krankheitsprogression hemmen würden.
Ich habe gelesen, dass Pa-tient:innen mit ATTR-Amyloidose nach Diagnosestellung durchschnittlich noch 2 Jahre leben. Stimmt das?
Diese Angaben beziehen sich auf Daten therapienaiver Patient:innen und eine Zeit, in der die Diagnose nur bioptisch gestellt werden konnte und daher oft sehr spät gestellt wurde. Die heute verfügbaren Therapien ermöglichen Patient:innen einen stabilen Verlauf über viele Jahre. Je früher im Krankheitsverlauf die Diagnose gestellt wird, umso besser ist die Prognose. Nimmt man ATTR-Patient:innen aller Stadien zusammen, leben nach 5 Jahren noch 50 %.
Wissenswertes für die Praxis
Ein- oder beidseitiges Karpaltunnelsyndrom bzw. Z. n. Operation eines solchen verbunden mit Septumdicke ≥ 15 mm bei fehlenden Hypertrophiezeichen im EKG sollen zu einer weiterführenden Abklärung hinsichtlich ATTR-Amyloidose führen.
Literatur bei den Verfasserinnen