


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Cystische Fibrose
Steigende Lebenserwartung dank neuer Therapien
2. Mai 2025
Cystische Fibrose (CF) ist die häufigste autosomal-rezessiv vererbte Erkrankung der „rare diseases“; laut Register sind in Österreich ca. 800 Patient:innen betroffen. Die Erkrankung tritt auf, wenn beide Eltern eine mutierte Variante an einem Allel des CFTR-(Cystische-Fibrose-Transmembran-Regulator-)Gens am Chromosom 7 tragen und autosomal-rezessiv vererben. Etwa jeder 25. Mensch überträgt eine dieser Mutationen, ohne selbst krank zu sein; die Krankheitshäufigkeit liegt bei 1 : 3.500. Symptome können in allen sekretbildenden Organen auftreten, am stärksten sind die Lunge und der Gastrointestinaltrakt betroffen.
Ursache und Auswirkungen
Ein gestörter Salz-Wasser-Transport durch Fehlen oder Fehlfunktion der CFTR-Kanäle an der Zelloberfläche bewirkt eine erhöhte Viskosität des Schleims in der Lunge. Dadurch kommt es zu einer unzureichenden Clearance mit Keimbesiedelung und in weiterer Folge zum Lungenumbau und Verlust der Lungenfunktion. Die resultierende chronische Entzündung der Atemwege, Bronchiektasen und irreparable Lungenschäden bestimmen die Lebensqualität und vor allem die Lebenserwartung der Patient:innen (Abb.).
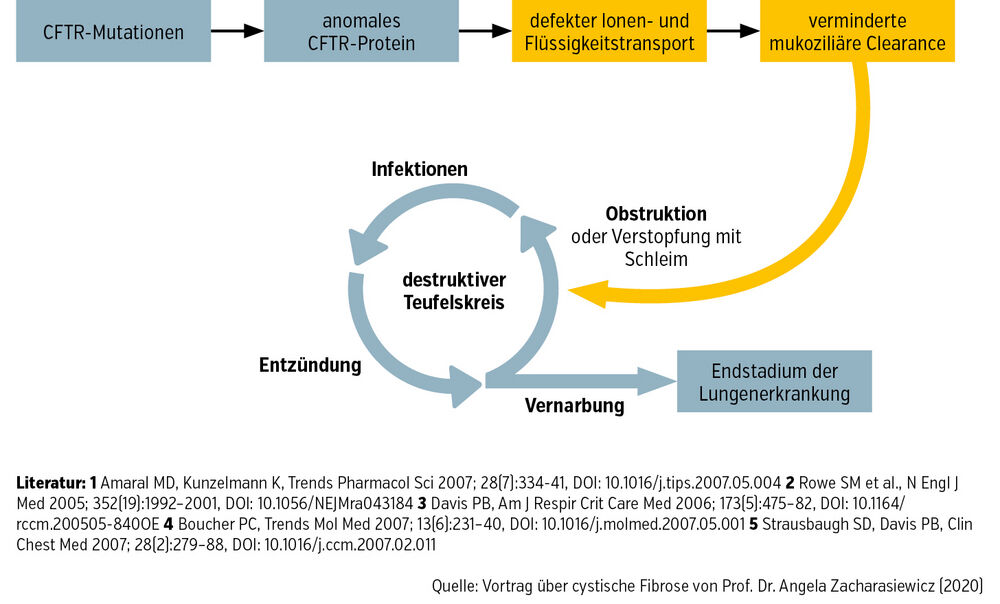
Abb.: Pulmonale Erkrankung aufgrund von CFTR-Protein-Fehlfunktion
Die Klasse-II-Mutation F508del verursacht als häufigste CF-Form (85 % der Patient:innen) zusätzlich eine exokrine Pankreasinsuffizienz. Folglich kann Fett nicht verwertet werden, was ohne Therapie zu Gedeihstörung, Untergewicht und erhöhter Infektanfälligkeit führt.
Diagnose und Therapie
Seit 1989 ist die CF genetisch entschlüsselt. Derzeit sind über 2.000 Mutationen bekannt, die in 6 Klassen eingeteilt werden. Die Diagnose kann nach pathologischem Neugeborenenscreening (erhöhtes immunreaktives Trypsinogen [IRT] + pankreasassoziiertes Protein [PAP]) früh gestellt werden und muss jedenfalls durch 2 positive Schweißtests in einem CF-Zentrum gesichert werden. Ein Chloridanteil von > 60 mmol/l im Schweißtest ist als pathologisch anzusehen.
„Die laufende medizinische Weiterentwicklung der kausalen Therapie erlaubt große Hoffnung für die therapeutische Zukunft.“
Nach bestätigter Diagnose erfolgt unmittelbar die Aufklärung der Eltern, und es wird im Rahmen der Erstuntersuchungen ein genetischer Test des CFTR-Gens durchgeführt. Die multidisziplinäre Betreuung erfolgt in regelmäßigen Abständen im CF-Zentrum. Ziel ist, die Lungenfunktion so gut wie möglich zu erhalten. Das gelingt nur, wenn wir als Betreuer:innen die Eltern und Patient:innen zu Expert:innen dieser bislang nicht heilbaren Erkrankung werden lassen!
Die bisher angewandte rein symptomatische Therapie umfasst:
- eine hochkalorische, fettreiche Ernährung mit Pankreasenzymersatz, Vitamin- und Salzsubstitution,
- tägliche Inhalationen mit höherprozentiger Salzlösung und Atemtherapie sowie Bewegungstherapie
- eine Antibiotikatherapie in hoher Dosierung für mindestens 2 Wochen bei speziellen Infektionen der Atemwege,
- bei schwerem Lungenfunktionsverlust Sauerstoffgabe und als letzte Option die Lungentransplantation.
Neue kausale Modulatortherapie
Seit 2020 gibt es sogar CFTR-Modulatoren für Patient:innen mit Klasse-II-Mutation und neu speziell die Triple-Therapie. Diese orale Therapie ermöglicht bzw. verbessert die Chloridkanalfunktion deutlich. Davon profitieren Patient:innen mit F508del durch eine merklich gesteigerte Lungenfunktion und weniger schwere Infekt-Exazerbationen. Obwohl die Pankreasfunktion nicht wiederherstellbar ist, kommt es zu besserer Gewichtszunahme und zur Erhöhung der Lebensqualität. Die mittlere Lebenserwartung stieg innerhalb der letzten 5 Jahrzehnte von 22 auf über 60 Jahre.

Autorin:
OÄ Dr.in Claudia Mori
OÄ Dr.in Claudia Mori
Wiener Gesundheitsverbund, Klinik Ottakring, Abteilung für Kinder- und Jugendheilkunde, PAVILLON 6

Ursprünglich erschienen:
AEK 09|2025
AEK 09|2025
WISSENSWERTES FÜR DIE PRAXIS
- Kinder mit Mekoniumileus, Gedeihstörungen oder chronischer Bronchitis sollen zum Schweißtest überwiesen werden.
- Bei Erwachsenen mit Polyposis nasi, Bronchiektasen oder Fertilitätsstörungen ist eine CF durch Schweißtest auszuschließen.
- CF-Patient:innen sollen entsprechend der Impfempfehlung immunisiert und zu Sport motiviert werden.
WAS PATIENT:INNEN WISSEN WOLLEN
Wann erhalte ich eine Modulatortherapie?
Bewilligung bei CF-Mutationen mit gesicherter Wirksamkeit, die Entscheidung treffen Sie gemeinsam mit Ihrem CF-Behandlungsteam.
Muss ich trotz Modulatoren weiterhin inhalieren?
Ja, unbedingt, da das Medikament nicht heilt, sondern nur die Kanalfunktion unterstützt. Die Inhalation bleibt essenziell für die Lungen-Clearance.
FACHÜBERGREIFENDE ZUSAMMENARBEIT
Die Zusammenarbeit mit niedergelassenen Kolleg:innen ist uns als Spezialist:innen für CF sehr wichtig! Für Fragen steht unser CF-Team, Pädiatrische Abteilung der Klinik Ottakring, gerne zur Verfügung. Terminvereinbarungen für Schweißtests telefonisch Montag bis Freitag unter: 01 491 50 28 18
Literatur bei der Verfasserin
Bildnachweis
Vorschaubild: © The 2R Artificiality – stock.adobe.com