
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Chronisch inflammatorische demyelinisierende Polyradikuloneuropathie
Wenn das Immunsystem die Nerven angreift
31. Oktober 2025
Die chronisch inflammatorische demyelinisierende Polyneuropathie (CIDP) verläuft meist progredient und ist häufig mit sensomotorischen Symptomen assoziiert.
Krankheitssteckbrief
Die CIDP ist eine entzündliche Erkrankung des peripheren Nervensystems. Die geschätzte Prävalenz liegt bei 1–9/100.000 Personen. Der Altersgipfel der Erkrankung liegt zwischen dem 40. und 60. Lebensjahr, sie kann jedoch grundsätzlich in allen Alterskategorien auftreten. Männer sind häufiger betroffen. Die Erkrankung kann anfänglich undulierend mit spontanen Remissionen verlaufen und entwickelt sich üblicherweise chronisch progressiv mit sensomotorischen Symptomen über einen Zeitraum von 8 Wochen.
Neben typischer CIDP existieren Varianten, die sich bezüglich Krankheitsverlauf, Verteilung der Defizite und u. a. durch Autoantikörper von der klassischen CIDP unterscheiden. Die CIDP gilt als eine der wichtigsten immunvermittelten Neuropathien, die bei rechtzeitiger Diagnosestellung gut behandelbar ist. Glukokortikoide und IVIG bleiben auch in den neuen Leitlinien Therapie der 1. Wahl.
Verlaufsformen, Varianten und Red Flags
Die neuen internationalen Leitlinien unterscheiden zwischen einer „typischen CIDP“ und „CIDP-Varianten“. Der Begriff „atypische CIDP“ ist obsolet. Die revidierten EFNS/PNS-Leitlinien unterscheiden nur noch eine „definitive CIDP“ und eine „mögliche CIDP“.
Typische CIDP
Die typische CIDP ist charakterisiert durch ein progredientes oder rezidivierendes, über mindestens 8 Wochen fortschreitendes Krankheitsbild mit distalen und proximalen sensomotorischen Symptomen. Dabei besteht entweder eine Areflexie oder Hyporeflexie an den betroffenen Extremitäten. Die Latenz bis zur Diagnosesicherung kann Jahre betragen, sodass Atrophien und/oder eine progrediente Gangstörung nicht selten sind.
In etwa 15 % der CIDP-Fälle tritt die Erkrankung akut mit raschem klinischem Verlauf auf, sodass das Plateau der Erkrankung innerhalb von 4 Wochen erreicht wird und daher einem Verlauf des Guillain-Barré-Syndroms (GBS) sehr ähnelt. Anders als beim GBS treten bei der „akuten CIDP“ (A-CIDP) nach 8 Wochen erneute Verschlechterungen auf. Beim GBS sind Fluktuationen im Erkrankungsverlauf innerhalb der 8 Wochen nicht außergewöhnlich. Die typische CIDP spricht üblicherweise gut auf die Immuntherapie an. Je nach klinischen Charakteristika lassen sich verschiedene Varianten unterscheiden (Tab.).
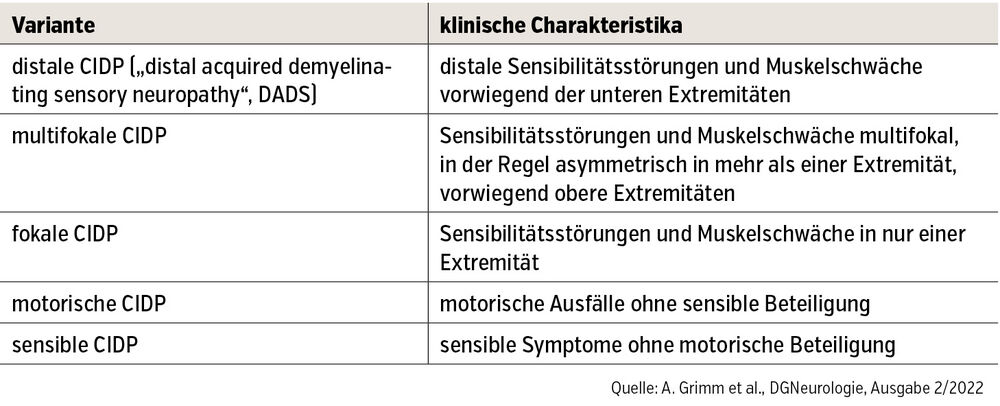
Tab.: CIDP-Varianten
Diabetes und CIDP
Wenngleich die diabetische Neuropathie mit ca. 30 % die häufigste Form der Polyneuropathie ist, scheint die CIDP unter Diabetespatient:innen häufiger aufzutreten. Mögliche Hinweise für die Komorbidität von Diabetes mellitus und CIDP sind a) rascher klinischer Verlauf trotz guter Diabeteseinstellung und b) schwerer klinischer Befall (z. B. generalisierte Areflexie, schwere Pallästhesiestörung, frühe Atrophie, fokale/asymmetrische Paresen).
(Para-)Nodopathien
Eine Unterform der Polyneuritis wird charakterisiert durch den Nachweis von Antikörpern gegen nodale oder paranodale Proteine (Anti-NF155-IgG, Anti-CNTN1-IgG, Anti-Caspr1-IgG und möglicherweise auch Anti-NF140/186-IgG). Diese Varianten scheinen eine eigene Krankheitsentität zu sein. Sie sprechen auf die übliche Immuntherapie der CIDP nicht gut an. Die Suche nach Autoantikörpern erscheint indiziert, wenn u. a. folgende Voraussetzungen vorliegen:
- fehlendes Therapieansprechen auf Standardtherapien (IVIG, Kortikosteroide)
- akuter oder subakuter fulminanter Beginn, frühere Diagnose von GBS oder A-CIDP
- niederfrequenter Tremor, Ataxie (ohne passende sensible Beteiligung oder andere Symptome einer Kleinhirnbeteiligung) oder eine überwiegend distale Schwäche
- assoziiertes nephrotisches Syndrom
Es gibt Hinweise, dass diese Patient:innen besser auf Rituximab ansprechen.
Diagnose
Majorkriterium zur Diagnosesicherung der CIDP ist der Nachweis der Demyelinisierung in der Elektroneurografie. Neu ist, dass die Demyelinisierung in sensiblen Nerven Eingang gefunden hat (verlängerte Latenz, reduzierte Amplitude, Minderung der Nervenleitgeschwindigkeit in mindestens 2 Nerven). Im klinischen Alltag werden in den Beinnerven nicht selten schwere axonale Schäden nachgewiesen, die durch die z. T. lange Latenz bis zur Diagnosesicherung erwartbar sind.
Daher empfiehlt es sich auch in diesen Fällen, die Armnerven neurophysiologisch zu untersuchen, auch wenn die Arme oligo- oder asymptomatisch sind. Häufig lassen sich proximale Demyelinisierungszeichen in Form von F-Wellen-Pathologie nachweisen.
Neben dem Nachweis der Demyelinisierung existieren ferner „supportive Kriterien“, die zur Unterstützung der CIDP-Diagnose hinzugezogen werden können:
- Bildgebung (MR-Neurografie, Nervensonografie)
- Liquorpunktion (entzündliches Liquorverlustsyndrom mit zytoalbuminärer Dissoziation)
- Nervenbiopsie
- Therapieansprechen auf die Immuntherapie
Behandlung
Die Therapie richtet sich nach Schweregrad und funktioneller Beeinträchtigung. Zentrale Säulen der Erstlinientherapie sind Glukokortikoide und intravenöse Immunglobuline (IVIG). Kortikosteroide werden bevorzugt in Form von Stoßtherapien eingesetzt, die oft besser verträglich sind als eine orale Dauertherapie. IVIG sind besonders bei motorischen Verlaufsformen wirksam und werden initial hochdosiert, danach in regelmäßigen Abständen zur Erhaltungstherapie gegeben. Je nach klinischem Verlauf kann das Therapieintervall individuell angepasst und verlängert werden, ggf. wäre auch eine Therapiepause zu diskutieren. Für die Erhaltungstherapie kann laut EAN/PNS-Leitlinien statt einer Behandlung mit IVIG auch auf subkutane Immunglobuline (SCIG) umgestellt werden, die von den Patient:innen selbst verabreicht werden. Dafür stehen in Österreich 2 unterschiedliche Präparate zur Verfügung: Ein SCIG, das wöchentlich verabreicht wird, sowie ein hyaluronidaseunterstütztes SCIG, das alle 3–4 Wochen verabreicht wird, was die Adhärenz vereinfacht.
Die Plasmapherese ist insbesondere bei fulminanten Verläufen oder Therapieversagen eine wirksame Option, wird jedoch nicht überall angeboten und ist mit periprozeduralen Risiken (u. a. Sepsis, Thrombose) verbunden. Bei inadäquatem Therapieansprechen oder Nebenwirkungen kann innerhalb der Initialtherapie die Substanz gewechselt werden (IVIG, Glukokortikoide, Plasmapherese), bevor eine duale Therapie in Betracht gezogen wird. Bei refraktären Verläufen oder speziellen Unterformen kommen Immunsuppressiva wie Azathioprin oder Mycophenolat sowie B-Zell-depletierende Therapien (z. B. Rituximab) zum Einsatz.
Mittlerweile steht mit der kürzlich erfolgten Zulassung von Efgartigimod alfa (subkutane Anwendung) eine weitere Therapieoption für Erwachsene mit progredienter oder rezidivierend aktiver CIDP zur Verfügung, sofern zuvor eine Behandlung mit Kortikosteroiden oder Immunglobulinen erfolgt ist.
Entscheidend ist eine individuelle, gemeinsam mit den Patient:innen getroffene Therapieentscheidung (Shared Decision Making). Ziel ist es, die Symptomprogression zu stoppen, die Funktion zu erhalten und die Lebensqualität nachhaltig zu verbessern.

Autor:
Prof. Dr. Min-Suk Yoon
Prof. Dr. Min-Suk Yoon
Klinik für Neurologie, Augusta-Kranken-Anstalt, Hattingen

Ursprünglich erschienen:
AEK SH Seltene Erkrankungen|2025
AEK SH Seltene Erkrankungen|2025
Literatur beim Verfasser
Dieser Beitrag ist ursprünglich in
Universum Innere Medizin 05/2025,
Themenheft Seltene Erkrankungen,
erschienen.
Universum Innere Medizin 05/2025,
Themenheft Seltene Erkrankungen,
erschienen.
Bildnachweis
Vorschaubild: © Katrin_Primak – stock.adobe.com