


























































































Die österreichische Fachzeitschrift für Rheumatologie mit wissenschaftlichen Updates zu Pathogenese, Diagnostik und Therapie sowie DFP-Fortbildung in jeder Ausgabe.
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
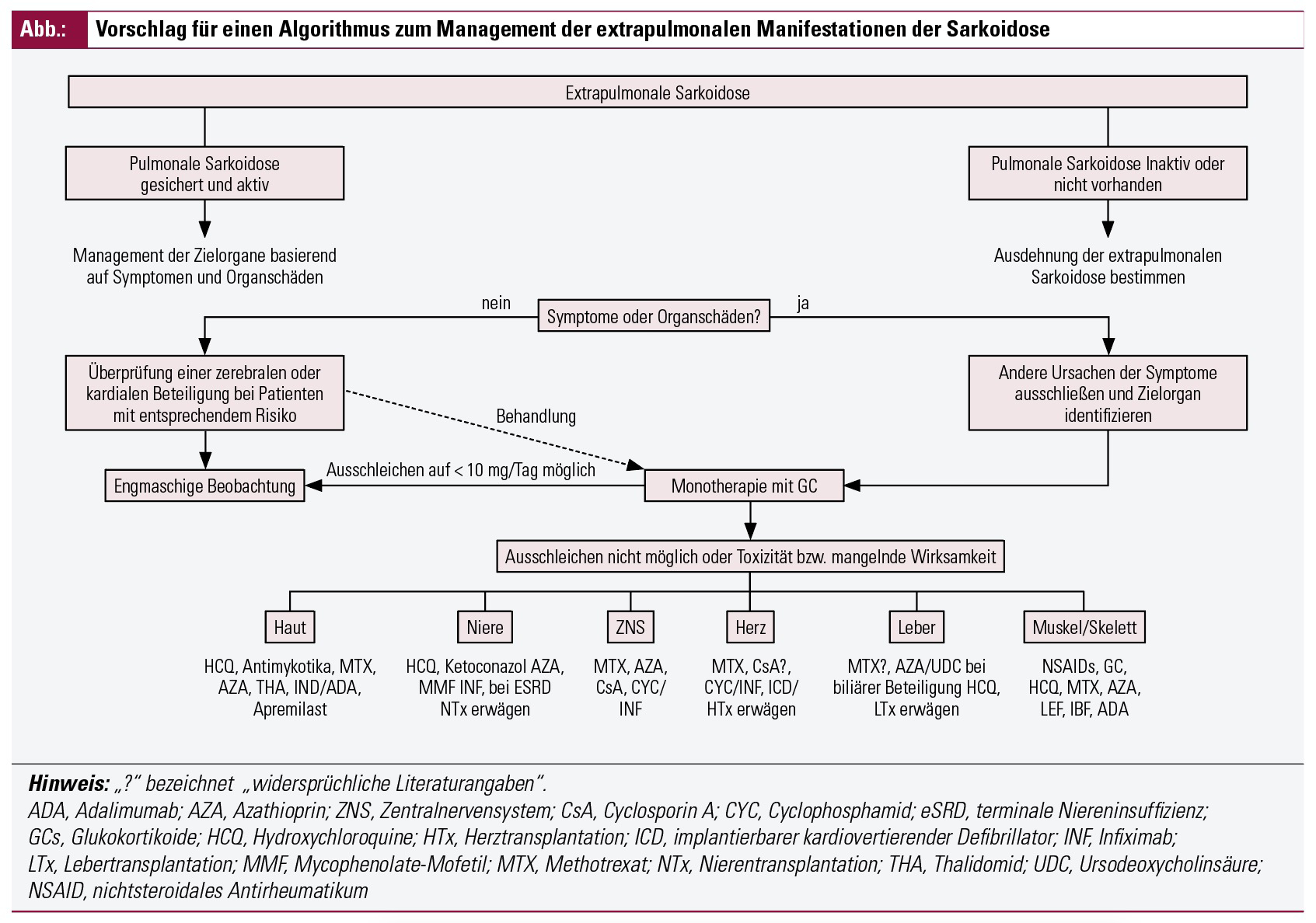
Die ACCESS-Studie hat gezeigt, dass von 736 untersuchten Sarkoidose-Patienten 50 % eine Kombination aus pulmonaler und extrapulmonaler Sarkoidose hatten. Eine isolierte extrapulmonale Sarkoidose kam hingegen nur bei 2 % der Betroffenen vor. Unter afroamerikanischen Studienteilnehmern war die Prävalenz der extrapulmonalen Sarkoidose höher als unter den kaukasischen. Obwohl sich bei fast zwei Drittel der Patienten nach ein bis zwei Jahren eine Remission einstellt, kommt es bei bis zu einem Drittel zu einem chronischen oder progressiven Krankheitsverlauf. Die Diagnosestellung erfordert den Ausschluss anderer Krankheitsursachen und basiert auf einer mit der Erkrankung vereinbaren medizinischen Vorgeschichte, klinischen Befunden, typischer Bildgebung und dem histologischen Nachweis nichtverkäsender Granulome. Bei einer histologisch gesicherten pulmonalen Sarkoidose kann bei typischer radiologischer Morphologie und nach Ausschluss anderer Ursache oft auf eine Biopsie des extrapulmonalen Organs verzichtet werden. Ein möglicher Zugang ist in der Abbildung dargestellt.
Bei 9–37 % der Patienten mit extrapulmonaler Sarkoidose treten Hautläsionen auf. Der Befall der Haut durch eine großknotige Sarkoidose wird als Lupus pernio (von lat. lupus = Wolf und pernio = Frostbeule) bezeichnet. Es finden sich flächenhafte, livide Infiltrationen von Nase, Wangen und Ohrläppchen. Bei der kleinknotigen Sarkoidose finden sich papulöse bis erbsengroße oder auch kleinknotige Hautveränderungen im Gesicht, an den Extremitäten, am Rumpf und an der Schleimhaut.
Hautläsionen bedürfen nur dann einer Behandlung, wenn sie für die Betroffenen ein schweres kosmetisches Problem oder eine psychische Belastung darstellen beziehungsweise nichtkontrollierbar sind. Die Erstlinientherapie für dicke plaqueförmige Läsionen ist eine lokale Glukokortikoidtherapie, bei der das Glukokortikoid direkt in die Läsionen injiziert wird. Bei progredienten Läsionen ist eine systemische Therapie, meist mit Glukokortikoiden oder Hydroxychloroquin, notwendig.
Unabhängig von dieser kutanen Manifestation hat ca. einer von fünf Patienten mit Sarkoidose ein Erythema nodosum. Das Erythema nodosum tritt häufig bei Patienten mit Löfgren-Syndrom auf, der akuten Form der Sarkoidose, welche durch bihiläre Lymphadenopathie und (Peri-)Arthritis des Sprunggelenks gekennzeichnet ist. Für gewöhnlich ist das Auftreten eines Erythema nodosum beim Löfgren-Syndrom ein Hinweis für eine gute Prognose.
Eine isolierte Beteiligung der Niere kommt nur bei 5 % aller Fälle von extrapulmonaler Sarkoidose vor. Im Verlauf der Erkrankung dürfte es jedoch bei einem wesentlich größeren Anteil zu einer Nierenbeteiligung kommen. Sarkoidose kann die Niere auf verschiedene Art und Weise betreffen:
Zur Vermeidung schwerer chronischer Schädigungen der Nieren sollten alle Patienten, bei denen eine Sarkoidose diagnostiziert wurde, auf das Vorliegen einer möglichen renalen Beteiligung untersucht werden. Die Abklärung sollte eine Bestimmung von Serum-Kreatinin, Harnstoff-Stickstoff, eGFR, Serumprotein und -kalzium, Harnprotein und -kalzium, Erythrozyten und Leukozyten im Harnsediment, 25-Hydroxyvitamin D3 sowie 1,25-Dihydroxyvitamin D3 umfassen. Liegt eine Hyperkalziämie oder Hyperkalziurie vor, kann eine PTH-Bestimmung Aufschluss über Ausmaß und Ursache der Deregulation des Kalziumspiegels geben. Goldstandard zur Diagnosesicherung einer renalen Sarkoidose bleibt die Nierenbiopsie.
Wichtigster Eckpfeiler der Behandlung einer interstitiellen Nephritis sind Glukokortikoide. Diese sind meistens sehr effektiv und wurden erfolgreich zur Behandlung von Hyperkalziämie und Hyperkalziurie eingesetzt. In vielen Fällen ist zur Normalisierung des Kalziumspiegels jedoch ein Absetzen von Vitamin-D- Ergänzungspräparaten ausreichend. Bei Patienten, bei denen Glukokortikoide nicht zum Erfolg führten, wurde Ketoconazol oder Hydroxychloroquin erfolgreich eingesetzt. Außerdem wurden – wenn auch nicht sehr häufig – bei fehlendem Ansprechen auf Glukokortikoide zusätzlich Immunsuppressiva wie Azathioprin (AZA) oder Mycophenolat-Mofetil (MMF) zur Behandlung der granulomatösen interstitiellen Nephritis verwendet.
Kommt es aufgrund einer therapierefraktären renalen Sarkoidose oder eines Therapieversagens zum terminalen Nierenversagen, besteht die Möglichkeit einer Nierentransplantation.
Eine Beteiligung des ZNS oder des peripheren Nervensystems stellte eine der schwereren Komplikationen der Sarkoidose dar und kommt bei 5–15 % aller Patienten vor. Bei 50–70 % der Fälle mit Neurosarkoidose sind Symptome, die durch Beeinträchtigung der Hirnnerven, des Hypothalamus oder der Hypophyse entstehen, die klinische Erstmanifestation.
Der Goldstandard zur Diagnosesicherung einer Neurosarkoidose ist eine ZNS-Biopsie mit Nachweis der typischen Granulome. Da die histologische Diagnose der Sarkoidose oftmals bereits durch Biopsien anderer betroffener Stellen gesichert ist, kann die Diagnose einer Neurosarkoidose in der Regel auch ohne ZNS-Biopsie gestellt werden.
Häufigste neurologische Manifestation der Erkrankung ist eine uni- oder bilaterale kraniale Neuropathie der Gesichtsnerven oder des Nervus opticus. Mögliche Ursachen für die Parese der Hirnnerven sind Granulome der Nerven, erhöhter intrakranialer Druck oder granulomatöse basale Meningitis. Mögliche Symptome sind Mononeuritis multiplex, Guillain- Barré-ähnliche Syndrome, Polyradikulopathie oder Polyneuropathie.
Da es schwierig ist zu biopsieren, ohne Schäden zu verursachen, ist die definitive Diagnosestellung einer Neurosarkoidose eine Herausforderung. Die sensitivste nichtinvasive Methode zur Diagnosesicherung ist das MRT, allerdings mangelt es an Spezifität.
Besteht bei Patienten, die eine bestätigte systemische Sarkoidose haben, der Verdacht einer Beteiligung des ZNS, sollte sofort mit einer Behandlung begonnen werden, da Neurosarkoidose mit einer hohen Mortalität und Morbidität einhergehen kann. Therapie der ersten Wahl sind Glukokortikoide, typischerweise mit einer initialen Dosierung von 1 mg/kg/Tag. In schweren Fällen kann eine Stoßtherapie mit 500–1.000 mg Methylprednisolon/Tag über drei Tage mit anschließendem oralem Ausschleichen gegeben werden. Bei refraktärer Erkrankung oder erheblichen Nebenwirkungen der Glukokortikoide wurden auch Methotrexat (MTX), Azathioprin (AZA), Cyclosporin A (CsA) und Cyclophosphamid (CYC) erfolgreich eingesetzt, allerdings handelt es sich hier mehr um Erfahrungswerte als um Evidenz. Für Patienten, die auf eine medikamentöse Therapie nicht ansprechen, kann als letzte Maßnahme eine Bestrahlungstherapie mit 20–25 Gy erwogen werden.
Eine kardiale Sarkoidose tritt selten isoliert auf, sondern ist meist begleitet von einer systemischen Sarkoidose. Es wird vermutet, dass eine okuläre Sarkoidose mit choroidaler Beteiligung auf eine Dysfunktion des Gefäßendothels hinweist und mit kardialer Sarkoidose einhergeht. Daher ist in diesem Fall eine Untersuchung einer möglichen kardialen Beteiligung erforderlich.
Das Ausmaß der durch die Sarkoidose verursachten kardialen Erkrankungen reicht vom Zufallsbefund bei asymptomatischen Patienten bis zu lebensbedrohlichen Erkrankungen mit plötzlichem Herztod. Typische Befunde sind Herzblock, ventrikuläre Tachykardie, nichtanhaltendes Kammerflimmern oder Herzinsuffizienz aufgrund von Kardiomyopathie.
Sowohl bei der Erstuntersuchung als auch bei Folgeuntersuchungen ist eine Anamnese wichtig, um Symptome festzustellen und gegebenenfalls weitere Untersuchungen einzuleiten. Jede Struktur und jede Schicht des Herzens kann betroffen sein. Je nachdem, ob z. B. das Endokard, die Herzklappen, das Myokard oder das Perikard betroffen sind, treten unterschiedliche Symptome auf, wie etwa Palpationen, Synkopen, Kurzatmigkeit, Brustschmerzen und periphere Ödeme.
Die Möglichkeit einer kardialen Sarkoidose sollte erwogen werden bei:
Wenngleich die Myokardbiopsie mit Nachweis der nichtverkäsenden Granolume der Goldstandard zur Diagnose einer kardialen Sarkoidose ist, kann mittels kardialer MRT inklusive gadoliniumhaltigem Kontrastmittel in den meisten Fällen die Diagnose gestellt bzw. ausgeschlossen werden. Ist ein MRT nicht möglich, kann auch eine FDG-PET zur Anwendung kommen.
Patienten mit durch Biopsie bestätigter oder klinisch diagnostizierter extrakardialer Sarkoidose und einem der folgenden Symptome oder Anzeichen einer kardialen Sarkoidose sollten eine kardiale MRT erhalten:
Patienten mit extrakardialer Sarkoidose ohne Symptome oder Anzeichen einer kardialen Sarkoidose sollten regelmäßig (z. B. jährlich) einer klinischen Untersuchung und einem EKG unterzogen werden, um die potenzielle Entwicklung einer kardialen Sarkoidose zu erkennen.
Die kardiale Sarkoidose erfordert aufgrund des Risikos des plötzlichen Herztodes und der fortschreitenden Herzinsuffizienz eine rasche Behandlung. Eine langfristige Verbesserung gab es bei der Therapie mit 6 mg MTX einmal wöchentlich zusätzlich zu Prednison versus Prednison allein. In einer retrospektiven Studie wurden andere Behandlungsmöglichkeiten wie MTX, CYC und CsA untersucht. Bei 10 von 11 Patienten mit kardialer Sarkoidose wurde damit eine Verbesserung erreicht. In Fallstudien wurde von Therapieerfolgen mit Infliximab berichtet. Zu beachten ist, dass TNF-α-Blocker bei Patienten mit fortgeschrittener Herzinsuffizienz (NYHA II oder NYHA III) kontraindiziert sind, da es zu einer Verschlechterung der Symptome kommen kann.
Die Implantation eines ICD wird empfohlen für Patienten mit:
Als Ultima Ratio kann eine Herztransplantation in Erwägung gezogen werden, da eine solche nach bisher vorliegenden Erkenntnissen mit keiner erhöhten Komplikationsrate verbunden ist.
Eine gastrointestinale Sarkoidose ist äußerst selten und kommt nur in 0–3,4 % aller Fälle vor. Diese Zahlen basieren auf Autopsiestudien. Eine Beteiligung der Leber ist dagegen sehr häufig zu beobachten. Sie wurde bei Autopsien in bis zu 80 % der Fälle festgestellt. Eine asymptomatische Erhöhung von Leberenzymen, vor allem der alkalischen Phosphatase, ist bei bekannter Sarkoidose eines der häufigsten klinischen Zeichen und tritt bei etwa einem Drittel der Patienten auf, wobei Afroamerikaner häufiger betroffen sind als Kaukasier.
Zu den klinischen Symptomen bei Leberbeteiligung zählen Hepatomegalie, Fatigue, Schmerzen im rechten Oberbauch mit Juckreiz (5–15 %), Fieber, Ikterus und Gewichtsverlust (< 5 %).
Betrifft die Sarkoidose den Gastrointestinaltrakt, zeigen sich Morbus-Crohn-ähnliche Symptome wie Völlegefühl, Gewichtsverlust, Enteropathie mit Proteinverlust, Obstruktionen oder gastrointestinale Blutungen.
Die Abklärung einer Sarkoidose des Gastrointestinaltrakts, der Leber oder der Milz kann endoskopisch oder mittels CT, MRT beziehungsweise Ultraschall (mit oder ohne Kontrastmittel) erfolgen. Anschließend sollte die Diagnose mittels Biopsie verifiziert werden.
Die Behandlung einer Sarkoidose der Leber oder der Milz ist nur dann erforderlich, wenn die Patienten Symptome aufweisen. Glukokortikoide können Größe der Leber und Milz ebenso reduzieren wie die Anzahl der Granulome, sodass sich die Organfunktion in gewissem Ausmaß verbessert. Bei Leberversagen wäre die orthotope Lebertransplantation erfolgreich.
Eine muskuloskelettale Beteiligung tritt in etwa 15–25 % aller Sarkoidosefälle auf.
Eine akute Arthrose in Zusammenhang mit einer Sarkoidose tritt am häufigsten beim Löfgren-Syndrom auf (bihiläre Lymphadenopathie, Erythema nodosum, beidseitige Schwellung der Sprunggelenke).
Chronische Arthritis in Zusammenhang mit einer Sarkoidose kann anderen Formen der entzündlichen Arthritis ähneln. Eine Daktylitis etwa – als typisches Zeichen der Psoriasisarthritis – kann auch bei Sarkoidose auftreten. Das Gleiche gilt für seronegative Spondylarthropathien.
Bildgebende Verfahren sind wichtige Werkzeuge zur Abklärung einer muskuloskelettalen Sarkoidose. Typischerweise wird eine konventionelle Röntgenuntersuchung durchgeführt, aber auch Sonografie, MRT und FDG-PET können hilfreich sein. Eine histopathologische Abklärung ist unbedingt erforderlich, da bei bildgebenden Verfahren keine ausreichende Unterscheidung zu anderen möglichen Ursachen wie Tuberkulose oder malignen Neubildungen gegeben ist.
Zur Therapie der akuten (Peri-)Arthritis, wie sie beim Löfgren-Syndrom auftritt, können unter Umständen NSAR (nichtsteroidale Antirheumatika) ausreichend sein. Bei bis zu zwei Drittel der Patienten wird allerdings eine zusätzliche Therapie mit Glukokortikoiden notwendig sein. Insgesamt ist die Prognose jedoch gut, in vielen Fällen kommt es bereits nach einigen Wochen bis Monaten zu einer vollständigen Remission.
Die chronische Arthritis kann mit Glukokortikoiden allein nicht immer unter Kontrolle gebracht werden. Sowohl Antimalariawirkstoffe als auch krankheitsmodifizierende Medikamente wurden schon erfolgreich eingesetzt, allerdings fehlen kontrollierte randomisierte Studien in dieser Indikation. Primär wird MTX eingesetzt, in schweren Fällen können auch TNF-α-Inhibitoren wie Infliximab oder Adalimumab verwendet werden.
Eine Beteiligung der Augen findet sich bei 25 % der Patienten mit Sarkoidose. Wenngleich jeder Bereich des Auges betroffen sein kann, sind Uveitis, Keratokonjunktivitis sicca und adnexaler Befall am häufigsten. Auch retinaler Befall ist möglich. Neben trockenen Augen und beeinträchtigtem Visus können auch Photophobie und Augenschmerzen auftreten. Die Diagnose basiert in der Regel auf einer extrapulmonal gesicherten Sarkoidose in Verbindung mit einem typischen ophthalmologischen Lokalbefund.
Ein Augenbefall ist eine klare Indikation zur systemischen Therapie. Wegen des Risikos für sekundäres Glaukom und Katarakt kommen bei der okulären Sarkoidose neben systemischen und topischen Glukokortikoiden auch biologische Therapien zur Anwendung.
Eine Übersicht der Therapien bei extrapulmonaler Sarkoidose findet sich in der Abbildung.
Literatur bei den Verfassern

2. Medizinische Abteilung mit Pneumologie, Karl Landsteiner Institut für Lungenforschung und Pneumologische Onkologie, Wilhelminenspital, Wien

2. Medizinische Abteilung mit Pneumologie, Karl Landsteiner Institut für Lungenforschung und Pneumologische Onkologie, Wilhelminenspital, Wien

2. Medizinische Abteilung mit Pneumologie, Karl Landsteiner Institut für Lungenforschung und Pneumologische Onkologie, Wilhelminenspital, Wien


