


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Die autosomal-rezessive axonale Neuropathie mit Neuromyotonie (ARAN-NM)
14. Dezember 2012
Hereditäre Neuropathien Charcot-Marie-Tooth (CMT) sind mit einer geschätzten Prävalenz von 40/100.000 relativ häufige Ursachen von vererbten neuromuskulären Erkrankungen. Die häufigsten Formen werden autosomal dominant oder X-chromosomal vererbt, rezessive Formen sind seltener. Derzeit sind mehr als 50 Gene bekannt, die eine CMT verursachen.
Die meisten dieser Neuropathien sind sensomotorische Neuropathien, die sich klinisch nicht wesentlich voneinander unterscheiden. Elektrophysiologisch werden sie in axonale, demyelinisierende und intermediäre Formen unterteilt. Erbgang und Elektrophysiologie bestimmen neben epidemiologischen Faktoren die weitere genetische Abklärung. Selten sind typisch klinische Zusatzsymptome vorhanden, die auf eine Mutation in einem bestimmten CMT-Gen hindeuten.
Neue Neuropathieform
Dieses Jahr wurde eine neue Form einer autosomal rezessiv vererbten, hereditären, axonalen Neuropathie entdeckt, die sich durch spezifische klinische und elektrophysiologische Merkmale der Neuromyotonie auszeichnet1.
Der Beginn der Erkrankung liegt zwischen dem 3. und 20. Lebensjahr. Die PatientInnen berichten über Gangunsicherheit, distale Schwäche der Beine und später auch der Arme, sensible Symptome stehen im Hintergrund. Der Achillessehnenreflex (ASR) fehlt, die anderen Muskeleigenreflexe sind abgeschwächt oder fehlen ebenso. Die PatientInnen berichten über Krämpfe in Armen und Beinen und ein Steifigkeitsgefühl. In der klinischen Untersuchung findet sich gelegentlich eine verlangsamte bzw. erschwerte Öffnung der Hand nach kräftigem Faustschluss. Eine Perkussionsmyotonie ist nicht nachweisbar. Die Neurographie zeigt eine axonale sensible und motorische Schädigung, im EMG findet man kurze hochfrequente Entladungen im Sinne einer Neuromyotonie (Abb.), aber auch myokyme Entladungen. Die Serum-CK kann leicht bis mäßig erhöht sein.
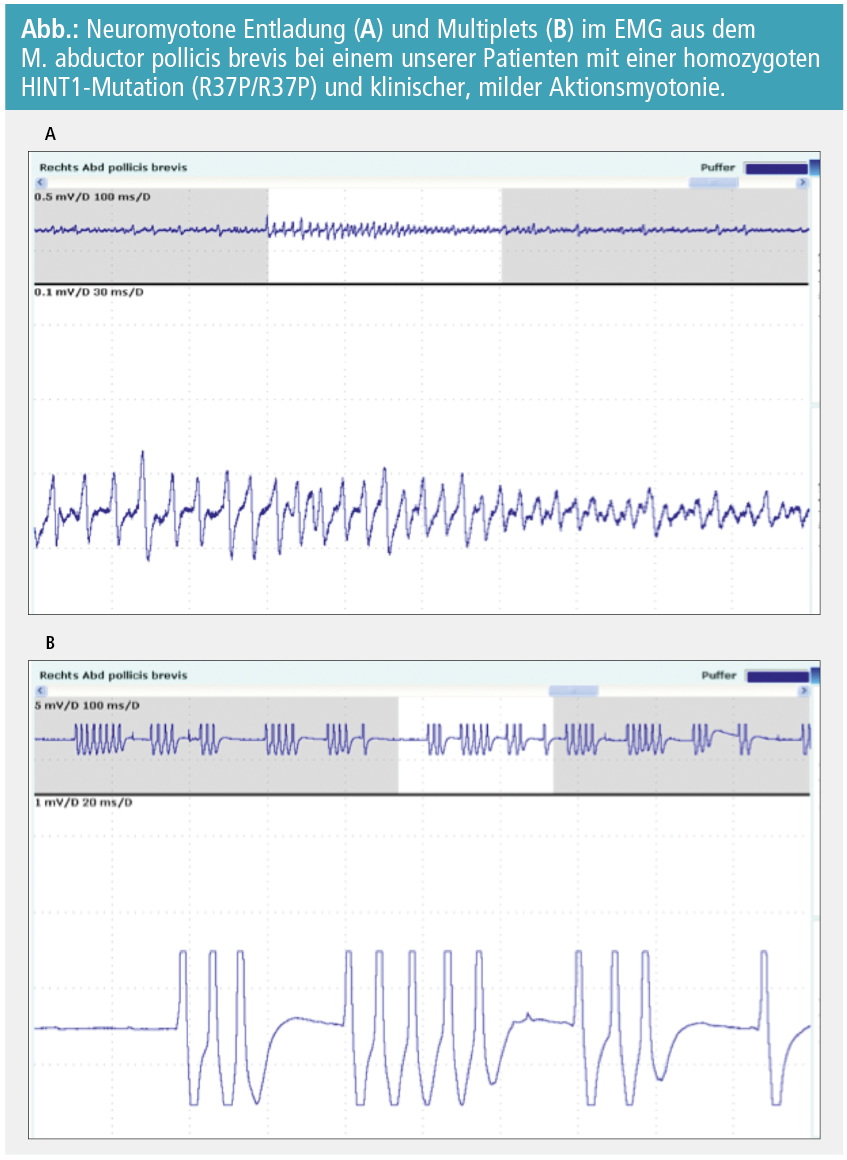
Nachdem rezessive Mutationen im HINT1-Gen zunächst unabhängig in Österreich und Belgien gefunden wurden, wurde eine Gruppe von 262 nicht verwandter PatientInnen mit axonaler rezessiver oder sporadischer Neuropathie auf HINT1-Mutationen untersucht. In 11 % der Fälle fanden sich homozygote bzw. compound-heterozygote Mutationen, die die Ursache der CMT-Erkrankung darstellen. In einer weiteren Untersuchung an einem spezifischen Subset von 31 PatientInnen mit einer axonalen rezessiven oder sporadischen Neuropathie mit Neuromyotonie wurden 21 PatientInnen mit HINT1-Mutationen gefunden, was für eine sehr starke Genotyp-Phänotyp-Korrelation spricht. In Österreich wurden bisher 4 Familien mit dieser neuen Neuropathie identifiziert1.
Derzeit sind 8 pathogene Mutationen im HINT1-Gen bekannt. Die Funktion des HINT1-Proteins ist derzeit nicht bekannt, aber es wird im N. ischiadicus der Maus stark exprimiert, sodass es doch eine Bedeutung für die Funktion peripherer Nerven haben dürfte. Funktionelle Studien haben auch gezeigt, dass die HINT1-Mutationen „Loss of function“-Mutationen sind. Die Rolle von HINT1-Mutationen in der Pathophysiologie der ARAN-NM ist allerdings noch unklar.
Kommentar: Diese Ergebnisse werden die genetische Diagnose von PatientInnen mit Neuropathien mit Neuromyotonie deutlich verändern. Denn bei diesem Phänotyp ist in Zukunft die Untersuchung des HINT1-Gens der erste Schritt. Es sollte bei allen sporadischen und rezessiven axonalen Neuropathien spezifisch nach myotonen Symptomen gefragt und auch eine EMG-Untersuchung durchgeführt werden, um nach neuromyotonen und myokymen Entladungen zu suchen. Der Nachweis neuromyotoner Entladungen und einer axonalen Neuropathie sind von Bedeutung, da die Kombination von Myotonie und distaler Schwäche fälschlicherweise den Verdacht auf eine myotone Dystrophie1 lenken könnte.
1 Zimon´ M et al., Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nature Genetics 2012; 44(10):1080–3.

AutorIn: Univ.-Prof. Dr. Wolfgang Löscher
Universitätsklinik für Neurologie,
Medizinische Universität Innsbruck
Zusammengestellt im Namen des Beirats „Neuromuskuläre Erkrankungen“

Univ.-Prof. Dr. Michaela Auer-Grumbach
Universitätsklinik
für Orthopädie, Medizinische Universität Wien
Zusammengestellt im Namen des Beirats „Neuromuskuläre Erkrankungen“

Ursprünglich erschienen:
neuro 04|2012
neuro 04|2012

