


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hirayama-Erkrankung – seltene Form einer Motoneuronerkrankung
28. September 2012
Epidemiologie: Über die Häufigkeit der HE liegen keine Daten vor. Im Allgemeinen scheint die Erkrankung in Japan weitaus häufiger zu sein als in Nordamerika oder Europa. Seit der Erstbeschreibung durch Keizo Hirayama im Jahre 1959 wurden in Japan bisher über 300 PatientInnen identifiziert, in Taiwan in einem 10-Jahres-Zeitraum 40 PatientInnen. In Österreich konnten wir in einer fast vollständigen Erhebung bisher 9 PatientInnen mit HE identifizieren. Die HE betrifft vorwiegend Männer. Das Verhältnis Männer zu Frauen wird mit 20 : 1 angegeben.
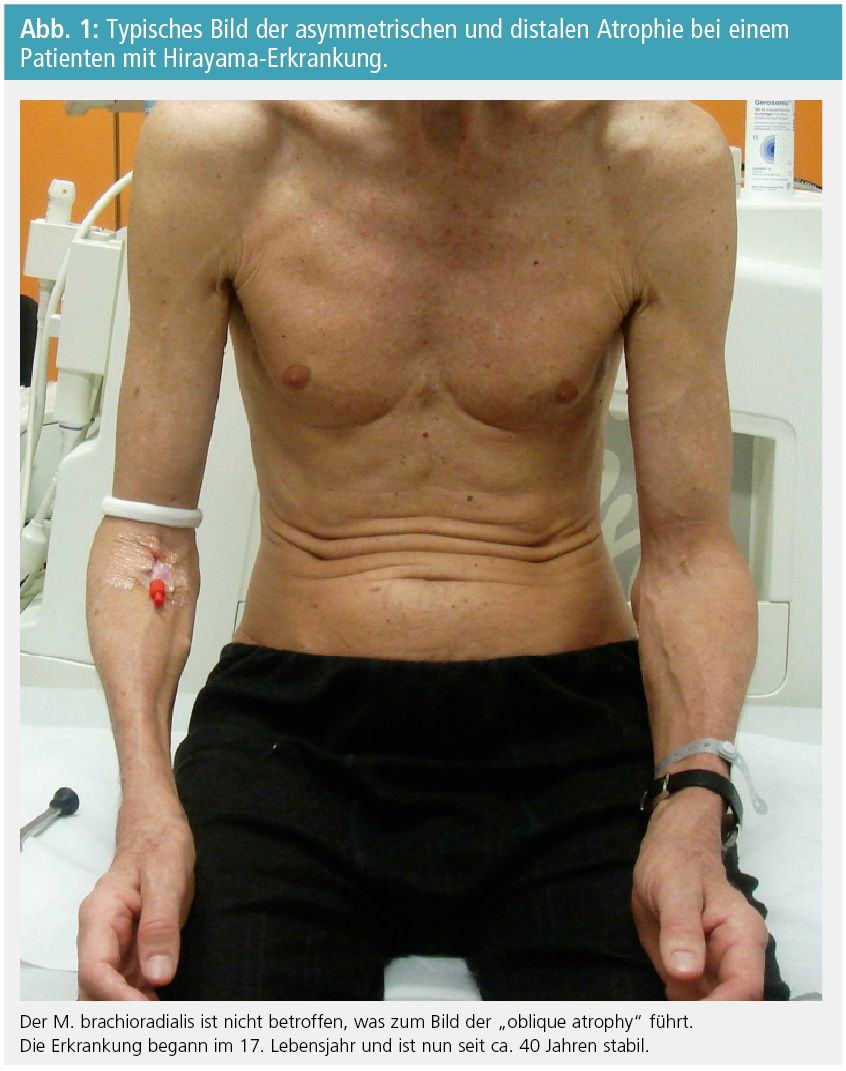
Klinische Präsentation: Der Beginn der Erkrankung liegt typischerweise in der Adoleszenz oder den frühen 20er-Jahren, in der Mehrzahl der Fälle zwischen dem 16. und 20. Lebensjahr. Die Symptome sind eine schmerzlose Schwäche und Muskelatrophie der distalen oberen Extremität. In der Regel ist nur eine Seite betroffen. Mit der Zeit kann gelegentlich auch die andere Seite, dann aber meistens schwächer, betroffen sein. Die Symptome sind typischerweise auf die intrinsische Handmuskulatur und Teile der Unterarmmuskulatur beschränkt und sparen klassischerweise den M. brachioradialis aus. Dies führt zum typischen Bild der „oblique amyotrophie“ (Abb. 1). Proximale Muskeln sind nur selten betroffen. Nach 1–5 Jahren kommt die Erkrankung zum Stillstand.
Weitere Charakteristika der Erkrankung sind die Kältelähmung und ein feinschlägiger Halte- und Aktionstremor. Eine Verschlechterung der Parese bei Kälte ist ein sehr typisches Zeichen, das sich fast bei allen PatientInnen findet. Der feinschlägige Haltetremor wurde auch als Minipolymyoklonus bezeichnet und findet sich ca. bei 2/3 aller Fälle. Die Muskeleigenreflexe an der oberen Extremität sind entweder normal oder abgeschwächt, aber auf keinen Fall gesteigert. Die Reflexe an der unteren Extremität sind unauffällig, ein positives Babinski-Zeichen findet sich ebenso wenig wie Blasenstörungen. Ebenso finden sich keine klinischen Zeichen einer Hirnnerven- oder Kleinhirnschädigung.
Diagnose: Die Diagnose HE basiert auf der klinisch-neurologischen Untersuchung, der Elektroneurographie, der Elektromyographie und dem zervikalen MRI in neutraler und Flexionsstellung. Die sensible Neurographie der Nerven am betroffenen Arm ist unauffällig, die motorische Neurographie zeigt amplitudenreduzierte Summenaktionspotenziale bei im Wesentlichen normalen Nervenleitgeschwindigkeiten. Die Nadelmyographie zeigt Zeichen der chronischen Denervation klinisch oder subklinisch betroffener Muskeln mit deutlich überhöhten und verbreiterten Muskelaktionspotenzialen, meist ohne Denervierungszeichen. Polyphasische Potenziale fehlen üblicherweise.
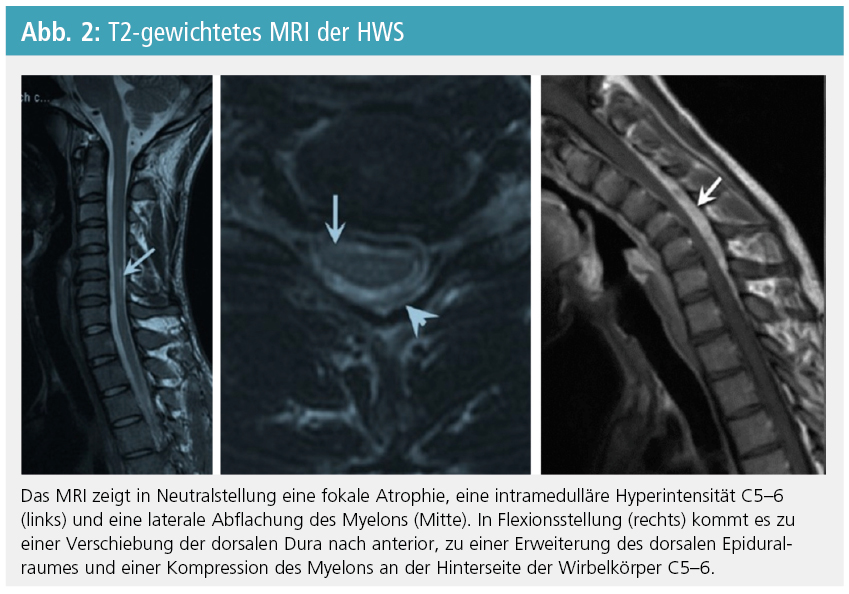
Das zervikale MRI in Neutralstellung zeigt eine fokale Atrophie des unteren zervikalen Myelons, eine abnormale Kurvatur, eine asymmetrische Abflachung des Myelons, intramedulläre Hyperintensitäten im unteren Zervikalmark und einen Verlust der Anheftung des Duralsacks an den darunter liegenden Knochen (Abb. 2). Das zervikale MRI in Flexionsstellung zeigt typischerweise eine Verschiebung der dorsalen Dura nach anterior, gleichzeitig mit einer Erweiterung des dorsalen Epiduralraumes und eine Kompression des Myelons an der hinteren Oberfläche der Wirbelkörper C5–6. Der erweiterte Epiduralraum stellt sich als halbmondfömige, hyperintense, kontrastmittelaufnehmende Masse dar, die in Neutralstellung verschwindet und dem posterioren Venenplexus entspricht (Abb. 2). Fokale Atrophie des Myelons, asymmetrische Abflachung des Myelons und fehlende Anhaftung haben gemeinsam eine diagnostische Treffsicherheit von 80 %. Wegen des Verzichts auf ein MRI in Flexionsstellung bleibt die HE häufig unentdeckt. Der Liquor ist bei HE unauffällig.
Differenzialdiagnosen: Differenzialdiagnosen der HE umfassen die Syringomyelie, die amyotrophe Lateralsklerose (ALS), die zervikale Myelopathie, Tumoren des Myelons und die traumatische Myelopathie.
Therapie: Die Therapie der HE ist empirisch und beschränkt auf Physiotherapie und die Verwendung einer Halskrause. Die Einschränkung der Nackenflexion durch eine Halskrause kann die weitere Progredienz der HE unterbinden. In ausgewählten Fällen führten Duroplastie (Vergrößerung der posterioren Fossa), anteriore zervikale Dekompression oder Rekonstruktion durch Sehnenverlagerung zu einem positiven therapeutischen Effekt.
Zusammenfassung: Die HE ist eine seltene Erkrankung der Motoneurone des unteren zervikalen Myelons und betrifft klinisch ausschließlich die oberen Extremitäten. Sie ist bedingt durch eine Dehnung des Myelons und Abhebung der dorsalen Dura vom darunter liegenden Knochen auf Grund einer unterschiedlichen Wachstumsgeschwindigkeit von Skelett und zervikalem Myelon und seinen Hüllen. Bei Anteflexion kommt es sekundär zu einer Kompression des unteren Halsmarks. Das Outcome der HE ist gut, da die aus der Kompression resultierenden Paresen und Atrophien nicht progredient sind und die Funktionalität der oberen Extremitäten meist erhalten bleibt.
Weiterführende Literatur:
- Hirayama K, Juvenile muscular atrophy of distal upper extremity (Hirayama disease). Intern Med 2000; 39(4):283–290.
- Hou C, Han H, Yang X, Xu X, Gao H, Fan D, Fu Y et al., How does the neck flexion affect the cervical MRI features of Hirayama disease? Neurol Sci 2012; doi: 10.1007/s10072-011-0912-x
- Huang YC, Ro LS, Chang H, Chen CM, Wu YR, Lee JD, Lyu RK, A clinical study of Hirayama disease in Taiwan. Muscle Nerve 2008; 37(5):576–582.
- Misra UK, Kalita J, Mishra VN, Kesari A, Mittal B, A clinical, magnetic resonance imaging, and survival motor neuron gene deletion study of Hirayama disease. Arch Neurol 2005; 62(1):120–123.
- Misra UK, Kalita J, Mishra VN, Phadke RV, Hadique A, Effect of neck flexion on F wave, somatosensory evoked potentials, and magnetic resonance imaging in Hirayama disease. J Neurol Neurosurg Psychiatry 2006; 77(5):695–698.
- Tashiro K, Kikuchi S, Itoyama Y, Tokumaru Y, Sobue G, Mukai E, Akiguchi I et al., Nationwide survey of juvenile muscular atrophy of distal upper extremity (Hirayama disease) in Japan. Amyotroph Lateral Scler 2006; 7(1):38–45.

Ursprünglich erschienen:
neuro 03|2012
neuro 03|2012

