


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Neurologische Symptome im Rahmen von paraneoplastischen Syndrome n
14. Dezember 2012
Diese Zusammenfassung zu paraneoplastischen neurologischen Syndromen (PNS) geht symptomorientiert vor und unterscheidet zwischen Hirnnervenstörungen, mentalen und anderen neurologischen Symptomen.
Antineuronale Antikörper: In den letzten Jahren entwickelte sich das Wissen über neuroimmunologisch ausgelöste PNS weiter. Ausgehend von zwei Krankheitsbildern, bei denen eine direkte Auswirkung der Antikörper (AK) nachgewiesen wurde, wie bei der Myasthenia gravis und dem Lambert-Eaton-Syndrom (LEMS), wurden zahlreiche AK-Assoziationen beschrieben. Eine Klassifikation von Graus2 teilt AK im Wesentlichen in onkoneuronale AK gegen intrazelluläre Antigene und Oberflächen- oder „Surface“-AK ein. Während bei den PNS, die durch onkoneuronale AK ausgelöst sind, Therapien schlecht ansprechen, sind PNS im Rahmen von Oberflächen-AK besser behandelbar. Bei den „klassischen“ onkoneuronalen AK (z. B. Hu, Yo, Ri) ergeben sich zunehmend Hinweise für einen T-Zell-mediierten Prozess3.
Zumindest 3 Gruppen von PNS werden aber in den konventionellen Einteilungen nicht berücksichtigt:
- PNS ohne nachweisbare oder mit atypischen AK: Diese Gruppe ist eine wichtige Quelle der Entwicklung bei der Erforschung der PNS,
- endokrine und humorale PNS4,
- PNS, bei denen deutliche Veränderungen ohne bisher erkennbaren kausalen Zusammenhang auftreten, wie beispiels-weise die Tumorkachexie, die sich deutlich von der Sarkopenie bei verminderter Nahrungsaufnahme unterscheidet.
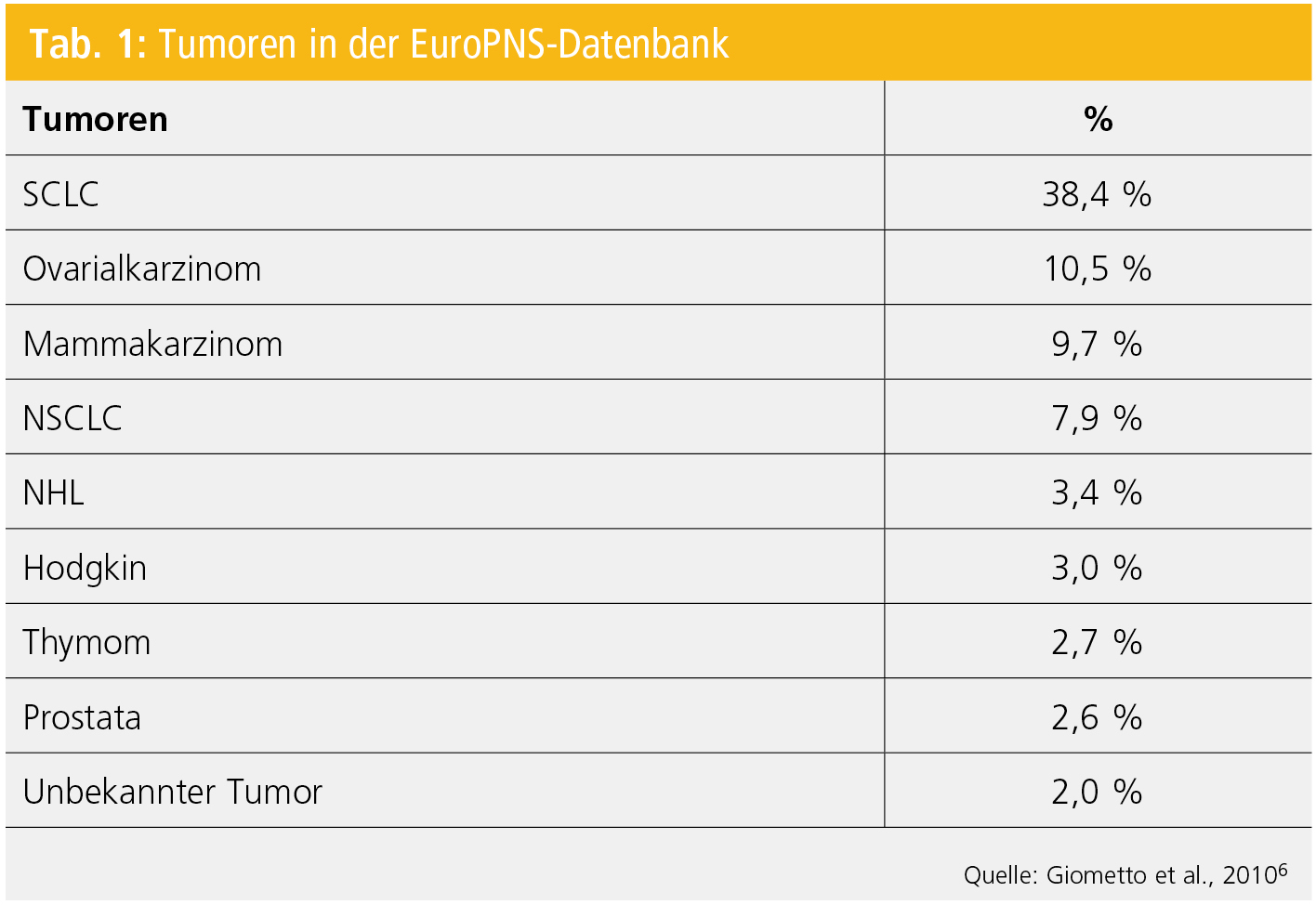
Tumoren: Obwohl PNS bei fast allen Tumoren beschrieben wurden, lässt sich eine Häufung bei kleinzelligen Lungenkarzinomen und bei gynäkologischen Tumoren feststellen. Eine EFNS-Guideline hat sich mit rationaler Tumorsuche bei vermuteten PNS beschäftigt5. In der EuroPNS-Datenbank konnte diese Verteilung bestätigt werden (Tab. 1)6.
Klinische Symptome: Was ein „klassisches“ oder wahrscheinliches PNS ist, ist das Ergebnis klinischer Untersuchungen und Beurteilungen. Ein Konsensus der EuroPNS7 teilte in sichere und wahrscheinliche PNS ein. Diese Einteilung ist zwar robust, berücksichtigt aber noch nicht die Oberflächen-AK, die erst in den letzten Jahren dazugekommen sind. Eine rezente Klassifikation der PNS anhand der AK wurde von Zuliani et al.8 publiziert.
Klinisch sind PNS oft charakteristisch, aber nicht spezifisch, d. h. idente neurologische Syndrome können auch bei immunologisch bedingten Krankheitsbildern ohne Neoplasma vorkommen: Beispiele sind die limbische Enzephalitis (LEMS) u. a.
Wahrscheinlichkeit: Die Wahrscheinlichkeit eines paraneoplastischen Syndroms liegt beim LEMS und bei der paraneoplastischen zerebellären Degeneration (PCD) bei ca. 50 %, bei der SSN bei 20–30 %, beim Opsoklonus-Myoklonus-Syndrom bei 20 %9.
PNS und klinische Symptome
In dieser Übersicht werden Symptome, Zeichen und Differenzialdiagnosen bei PatientInnen mit PNS dargestellt.
Hirnnerven
Sehstörungen kommen bei PNS selten vor. Ein Syndrom der PNS-Schädigung durch CAR-AK, vorwiegend gegen Recoverin, ist beschrieben. Die Symptome sind Visusverlust an beiden Augen, Photopsien, periphere Skotome (ringartig) und „Nachtblindheit“ als vorausgehende Störung. Das Krankheitsbild tritt vorwiegend bei Lungenkarzinomen auf und führt zu einer Schädigung der Sehnerven oder der Retina. Die CAR-bedingten Visusstörungen müssen gegenüber Prozessen im Auge (Lymphom, Metastasen) oder im Verlauf der Sehbahn abgegrenzt werden. Das Krankheitsbild tritt bei Lungentumoren, selten auch bei Melanomen auf.
Wesentlich häufiger ist undeutliches oder „unscharfes“ Sehen, was durch geringe Divergenzen der Bulbusachsen (Läsion im Hirnstamm, Hirnnervenverlauf, Orbita) auftreten kann und vorwiegend durch neoplastische Prozesse verursacht wird.
Sehunschärfe und Bulbusbewegungsstörungen sind bei pontinem Befall beim PNS der Hirnstammenzephalitis beschrieben10. Das ist ein sehr seltenes Geschehen, und in erster Linie müssen strukturelle Prozesse (Hirnstamm, Meningealkarzinomatose) ausgeschlossen werden. Assoziiert mit den Hirnstammsyndromen kann es zu Hypoventilation bis zur Apnoe kommen.
Das Opsoklonus-Myoklonus-Syndrom (OPM), auch „Dancing Eyes“-Syndrom genannt, ist durch irreguläre und ruckartige Augenbewegungsstörungen gekennzeichnet. Zusätzlich kommt es mehr oder minder ausgeprägt zu Myoklonien am Körper. Das OPM kommt bei zahlreichen Tumoren und bei Ri-AK vor. Es ist aber unspezifisch und kann auch parainfektiös auftreten.
Beim LEMS können inkomplette Ptosen und auch Sehunschärfe bis zur Diplopie auftreten. Charakteristisch sind autonome Beschwerden wie trockene Augen und trockener Mund.
Das „Numb Chin“-Syndrom beschreibt eine Sensibilitätsstörung im Austrittsbereich des N. mentalis. Das ist möglicherweise eine PNS, kann aber auch bei Metastasen im Verlauf des Nerven in der Mandibel oder bei Befall des Ganglion Gasseri vorkommen. Motorische Ausfälle des N. trigeminus, oft mit Schmerzen oder Sensibilitätsstörungen kombiniert, lassen an eine strukturelle Läsion des Nerven an der Schädelbasis denken.
Die kaudalen Hirnnerven können im Rahmen der paraneoplastischen Hirnstammenzephalitis betroffen sein, aber auch bei der PCD kommt es zu einer charakteristischen zerebellären Sprechweise. Sprechstörungen können auch bei Neuromyotonien vorkommen.
ZNS
Anfälle sind in der Neuroonkologie häufig. Es handelt sich vorwiegend um neoplastische, metabolische und auch toxische Prozesse, die als Ursache verschiedener Anfallstypen vorkommen. Bei PatientInnen mit limbischer Enzephalitis (LE) sind Anfälle sowohl beim Beginn des PNS als auch im Verlauf möglich.
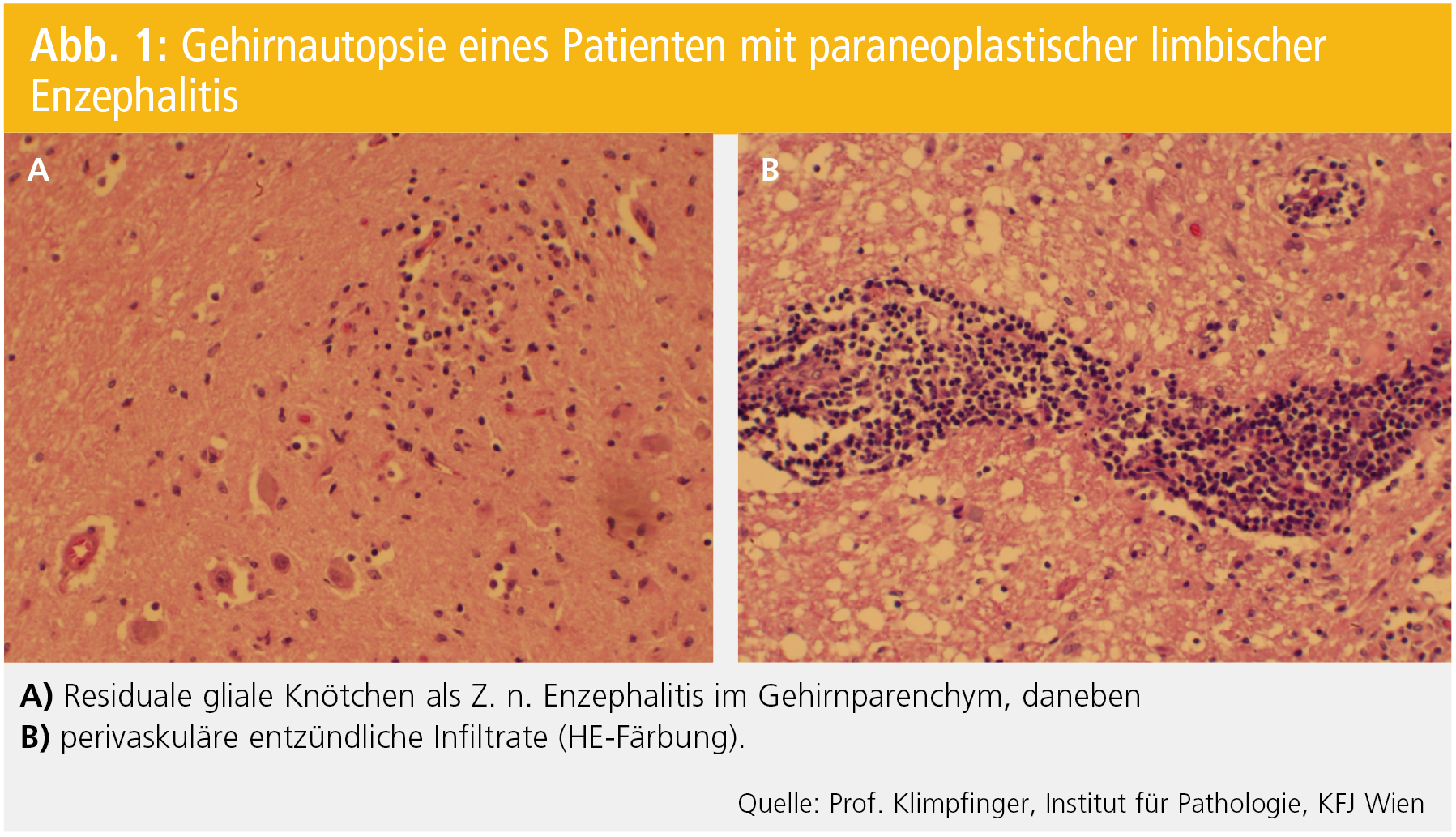
Mentale Veränderungen: Die bekannteste und auffälligste Störung ist die limbische Enzephalitis (LE). Diese ist durch oft akuten Beginn einer amnestischen Störung, möglicherweise auch durch Bewusstseinstrübung und Anfälle gekennzeichnet. Entzündliche Veränderungen unterschiedlichen Ausmaßes können im Hirnparenchym festgestellt werden (Abb. 1).
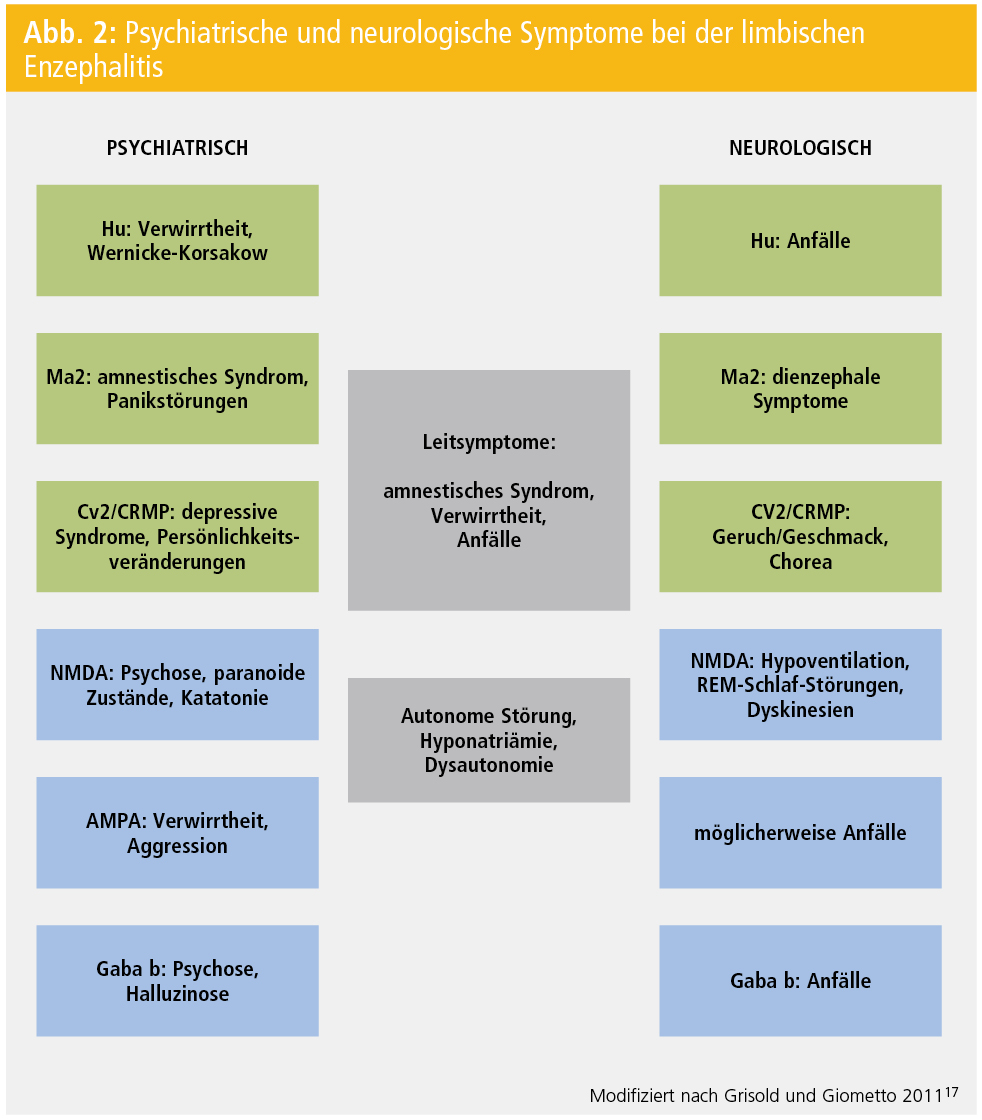
Phänomenologisch ähnelt die Psychopathologie und die Präsentation einem Wernicke-Korsakow-Syndrom. Die Ursache ist aber nicht einheitlich, denn die LE kommt bei onkoneuronalen AK (vorwiegend Hu) und bei Oberflächen-AK (VGKC, NMDA u. a.) vor. Auch idiopathische und nichtentzündliche Formen sind beschrieben. In der klinischen, psychiatrischen und neurologischen Manifestation werden Unterschiede beobachtet, die in der Abbildung 2 zusammengefasst werden.
Aus neurologischer und auch aus psychiatrischer Sicht sind immunmediierte Enzephalitiden in das differenzialdiagnostische Spektrum aufzunehmen, wobei besonders bei der NMDA-verursachten LE vorwiegend psychiatrische Symptome bis zur Katatonie auftreten.
Ataxie: Die paraneoplastische zerebelläre Degeneration (PCD) entwickelt sich akut oder subakut und stellt ein dramatisches Ereignis dar. Das Krankheitsbild erreicht rasch die volle Ausprägung und bleibt auf einem irreversiblen Plateau. Klinisch ist es ein „panzerebelläres“ Syndrom und beinhaltet Rumpf-, Stand- und Extremitätenataxie sowie zerebelläre Sprechweise und Schluckstörungen (Tab. 2). Selten wurden auch Verschlechterungen der PCD bei Tumorrezidiv festgestellt. Auch bei AK gegen Kalziumkanäle (VGCC) werden zerebelläre Störungen beschrieben.
Fast alle PCD treten zu Beginn der Tumorkrankheit auf, lediglich beim M.-Hodgkin-assoziierten Anti-Tr-Syndrom tritt die PCD später auf.
Toxische zerebelläre Syndrome können bei Gabe von hoch dosiertem 5-FU (5-Fluoruracil) und Cytosin-Arabinosid auftreten. Auch bei Intoxikationen mit Phenytoin können zerebelläre Symptome auftreten.

Motorik und EPS: PNS des ZNS betreffen die Willkürmotorik vorwiegend indirekt, da sensible Störungen und Koordinationsstörungen den Ablauf der Motorik negativ beeinflussen. Paresen im eigentlichen Sinn sind nicht zu erwarten. Extrapyramidale Störungen wie Parkinson-Syndrome und choreatische Störungen wurden in einer kleinen Zahl von PatientInnen in der EuroPNS-Datenbank beschrieben11.
Myelopathien: Myelopathien bei Tumoren sind selten, und in erster Linie von spinalen Kompressionen abzugrenzen. Ein seltenes und akutes Krankheitsbild ist die akute „transverse Myelitis“ (Querschnittsmyelitis), die zu akuten Querschnittsbildern führt. Isolierte Struktur- oder Strangdegenerationen können ebenso vorkommen.
Hypoventilation und Apnoe: Im Zusammenhang mit der limbischen Enzephalitis (Hu und NMDA) fielen mehrfach Hypoventilationen bis hin zur Apnoe auf. Eine Untersuchung der EuroPNS-Datenbank konnte diesen Verdacht beweisen und führt dies auf eine Hirnstammenzephalitis zurück10.
Hypothalamische Störungen: Funktionsstörungen wie Somnolenz, Hyperthermie, endokrine Störungen, Hyperhidrose und Myokymien werden in Zusammenhang mit Ma2, CRMP5 (CV2) und in Zusammenhang mit der limbischen Enzephalitis beschrieben.
Paraneoplastische endokrine Symptome
Diese treten als SIADH, Hyperkalziämie, Cushing-Syndrom (paraendokrine Hormonproduktion) und Hypoglykämie in Erscheinung4.
Autonomes System
Autonome Störungen liegen in unterschiedlicher Form vor. Die gastrointestinale Intussuszeption kann im Rahmen des Anti-Hu-Syndroms auftreten. Mundtrockenheit ist beim LEMS beschrieben. Andere, unspezifische autonome Störungen werden vorwiegend beim Anti-Hu-Syndrom beobachtet.
Peripheres Nervensystem
Paraneoplastische Polyneuropathien sind vorwiegend sensorisch. Immunmediierte Neuropathien wie das GBS und die CIDP sind sehr selten mit Tumoren assoziiert, wobei vorwiegend Lymphome beschrieben werden12. Mononeuropathien werden zwar in Einzelfällen beschrieben, systematische Untersuchungen fehlen mit Ausnahme des Karpaltunnelsyndroms, welches bei Paraproteinämien und Amyloidosen vermehrt auftreten kann. Höhergradige, vorwiegend motorische PNP sind bei Paraproteinämien, besonders beim POEMS-Syndrom13 beschrieben.
Myasthene Syndrome: Störungen des neuromuskulären Überganges können zu myasthenen Syndromen mit wechselnder Ausprägung führen. Besonders beim LEMS besteht eine vorwiegend proximale Schwäche mit geringer Hirnnervenbeteiligung und charakteristischerweise mit Areflexie (welche durch Fazilitierung – d. h. kurze maximale Muskelkontraktion oder repetitives Auslösen des Reflexes – unterbrochen werden kann), die eine Verwechslung mit Polyneuropathien wahrscheinlich macht. Neuromyotonien führen zu Paresen und „Muskelsteifheit“ sowie auch zu begleitenden Hyperhidrosen (Morvan-Syndrom).
Stiff-Person-Syndrome sind einerseits das „stiff person syndrome“, andererseits das „stiff limb syndrome“. Dabei kommt es einerseits zu paroxysmaler, oft durch Reize ausgelöster Muskelsteife andererseits zu bewegungsabhängiger Muskelsteife, die mit Rigidität oder Spastik verwechselt werden kann.
Myopathien: Akute Myopathien entsprechen fast immer dem typischen proximalen Muster. Als Ursache sind die Dermatomyositis (DM), wesentlich weniger die Polymyositis (PM) und die nekrotisierende Myopathie zu nennen. Bei der DM sind die begleitenden Hautveränderungen charakteristisch.
Proximale Muskelschwäche, vorwiegend mit Hautveränderungen, lässt an eine DM denken, während PM bei Tumoren seltener vorkommen. Bei den entzündlichen Myopathien bestehen oft Schluckstörungen, die anamnestisch hilfreich sind.
Bei nekrotisierenden Myopathien kann etwa in der Hälfte der Fälle (Angaben schwanken) bei Erwachsenen ein Tumorleiden vorliegen. Die nekrotisierende Myopathie ist durch proximale Schwäche und hohe CK-Werte gekennzeichnet und kann auch während eines Tumorleidens auftreten. Klinisch ähnelt das Krankheitsbild einer PM.
Iatrogen sind Myopathien bei chronischer Steroidtherapie abzugrenzen. Diese werden durch hohe Dosen von Kortison, z. B. Dexamethason, ausgelöst, die bei Hirntumoren und Hirnmetastasen eingesetzt und oft über lange Zeit als Dauertherapie angewendet werden. Neben dem Cushing-Syndrom sind Muskelschwäche der Hüft- und Oberschenkelmuskel (Hüftbeuger und Kniestrecker) prominent, Erstsymptom ist oft verminderte Kraft beim Aufstehen, z. B. von der Toilette. Klinisch kommt es oft zu drastischen Verschmächtigungen der Oberschenkelmuskeln. Selten können bei Vinca-Alkaloiden nekrotisierende Myopathien auftreten, hingegen scheint die durch Taxane ausgelöste proximale Myopathie häufiger zu sein. Myalgien werden bei der Therapie mit Taxanen und Gemcitabin beschrieben.
Tumorkachexie: Das wichtigste und häufigste PNS, nämlich die Tumorkachexie, ist weitgehend unerforscht. Sarkopenie der Skelettmuskulatur stellt möglicherweise auch einen prognostischen Faktor für das Überleben dar. Kriterien zur Unterscheidung zwischen Tumor- und Nahrungsmittelsarkopenie werden beschrieben. Es kommt zu Muskelatrophien im Rahmen einer Gewichtsabnahme. Die maximale Kraft bleibt noch sehr lange erhalten. Gelegentlich kann es bei der Perkussion zu einem Myödem kommen, die Reflexe sind fast immer erhalten14.
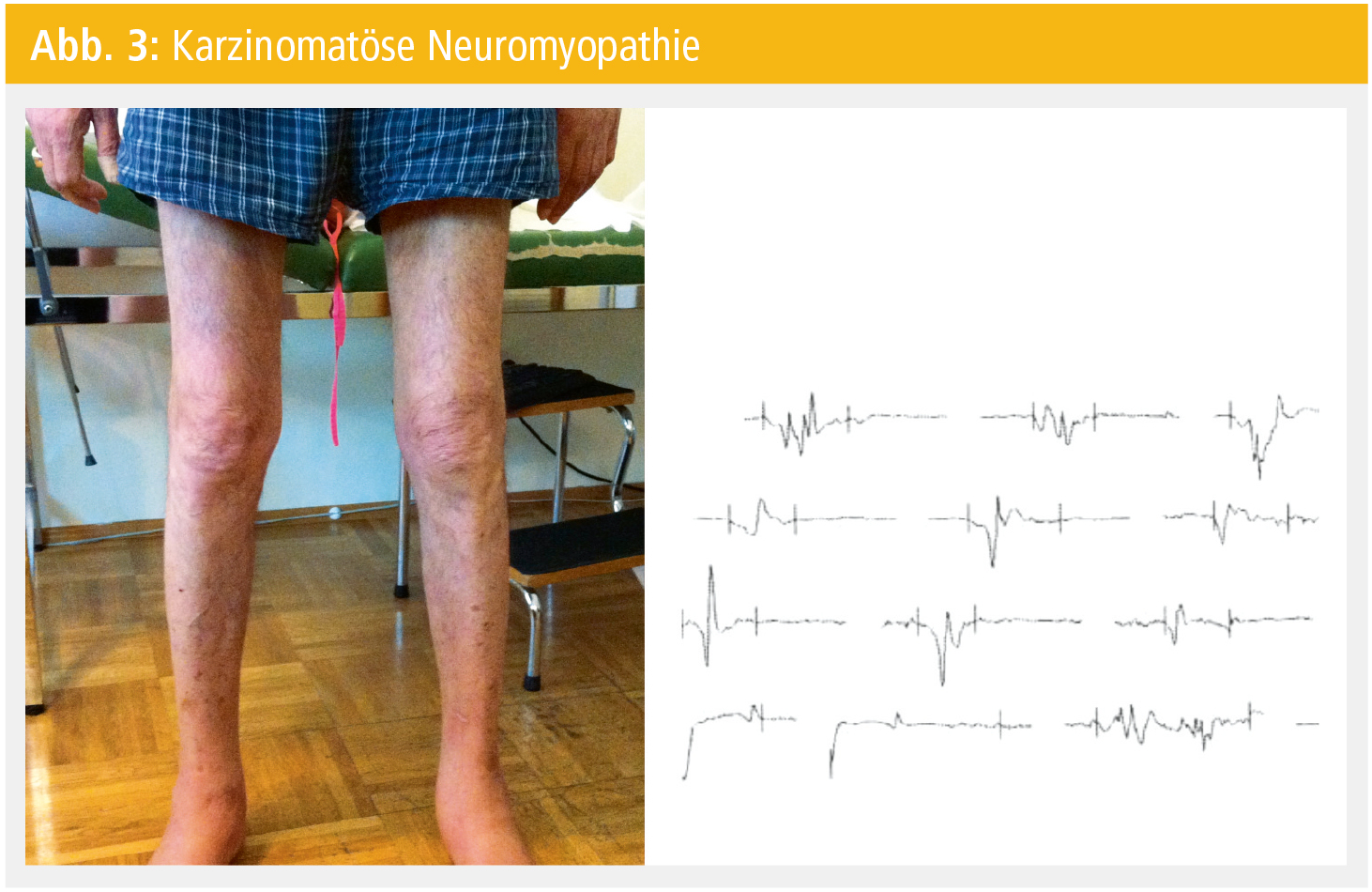
Eine früher beschriebene Form wurde als karzinomatöse Neuromyopathie bezeichnet, bei der hochgradige Atrophien mit relativ erhaltener Kraft und mit myopathischem EMG einhergehen können. (Abb. 3).
Bei Thymomen sind neben der Myasthenia gravis entzündliche Myopathien und sogenannte „rippling muscles“ beschrieben. Dabei kommt es zu wellenförmigen Muskelbewegungen, die für die PatientInnen sehr beeinträchtigend sind. Bemerkenswert ist, dass im EMG kein entsprechendes Korrelat gefunden werden kann.
Muskelkrämpfe in den Hand-, vorwiegend aber in Fußmuskeln weisen auf Denervation hin und sind gelegentlich Symptome einer sensomotorischen Neuropathie durch Chemotherapie (z. B. Vinca-Alkaloide).
Motoneuronerkrankungen: Die Frage, inwieweit das Krankheitsbild einer amyotrophen Lateralsklerose (ALS) im Rahmen eines Tumorleidens vermehrt vorkommt, ist eine jahrelange Kontroverse, die nicht entschieden ist. Zwei besondere Formen, das Bild der „lower motor neuron disease“ bei Paraproteinämien13, 15 und das Bild der primären Lateralsklerose (PLS) sind erwähnenswert. Bei der PLS kommt es zu einer Degeneration der Pyramidenbahnen mit Spastizität. Möglicherweise handelt es sich um eine Sonderform der ALS.
Sensibilität
Sensorische PNP und sensorische Ataxie: Die subakute sensorische Neuropathie (SSN) ist ein charakteristisches Krankheitsbild, bei der sensible Störungen, oft mit Schmerzen kombiniert, und eine beträchtliche sensorische Ataxie im Vordergrund stehen. Das Krankheitsbild beginnt subakut, typischerweise oft asymmetrisch an den oberen Extremitäten (Tab. 3). Manchmal kommt es auch zu Berührungsschmerzen mit temporaler Dispersion. Die Sensibilitätsstörung kann bis zur Unfähigkeit gehen, Gegenstände anzufassen, zu essen, zu stehen oder zu gehen. Objektiv besteht beim Finger-Nase-Versuch und beim Knie-Hacken-Versuch oft eine spinale Ataxie und auch eine spinale Pseudoathetose als Zeichen der sensiblen Deafferenzierung. Die SSN ist nicht PNS-spezifisch, sondern kommt auch beim Sjögren-Syndrom und idiopathisch vor.
Primäre sensorische Symptome kommen bei PNS des ZNS selten vor. Im Hirnnervenbereich können sensible Störungen im Trigeminusbereich („numb chin“) auftreten. Andere Gefühlsstörungen, wenn auch schlecht definiert, kommen bei der Neuromyotonie, besonders beim Morvan-Syndrom vor, auch beim LEMS werden distale sensible, schwer abgrenzbare Symptome beschrieben.

Spinale Sensibilitätsstörungen: Obwohl spinale Symptome oft im Zusammenhang mit paraneoplastischen Enzephalomyelopathien, insbesondere dem Anti-Hu-Syndrom beschrieben werden, sind Myelitiden im Rahmen von PNS eine Rarität. Die paraneoplastische Querschnittsmyelitis wird seit Jahren beschrieben, ist aber selten. Zusätzlich wird eine isolierte Hinterstrangdegeneration bei der SSN autoptisch beschrieben. Isolierte Hinterstrangdegenerationen bei PNS sind selten16 und können durch MR-Untersuchungen nachgewiesen werden.
Resümee
Die Entwicklung bei der Erforschung der PNS hat in den letzten Jahren zahlreiche neue Erkenntnisse gebracht. Das bezieht sich auf Klassifikation und Häufigkeit, den Nachweis der Oberflächen-AK und nunmehr den Nachweis zytotoxischer Mechanismen bei den Oberflächen-AK. Zusätzlich werden einige humorale, endokrine und auch ungeklärte PNS unterschiedlich akzeptiert.
Kenntnisse und diagnostische Möglichkeiten haben sich in den letzten Jahren deutlich verbessert, wobei das Hauptaugenmerk auf neuroimmunologisch bedingte Symptome in Assoziation mit AK gerichtet ist. Klinisch relevant für die Diagnose ist, dass die PNS fast immer vor dem Tumorleiden auftreten und dass zahlreiche PNS relativ spezifische Symptome aufweisen, die an ein PNS denken lassen müssen.
Klinische Syndrome des ZNS und des peripheren Nervensystems weisen zwar auf PNS hin, jedoch sind alle PNS unspezifisch, d. h. kommen auch auf immunologischer Basis ohne Tumorassoziation vor. Sowohl onkoneuronale als auch Oberflächen-AK sind sehr spezifisch, und die Untersuchung sollte möglichst von der klinischen Symptomatologie gesteuert werden. Screening nach PNS mit einem AK-Panel ist nicht sinnvoll.
Die AK – onkoneuronale AK und Oberflächen-AK – ergeben zusammen mit dem klinischen Syndrom und den Zusatzbefunden die definitive Diagnose und damit einerseits den Nachweis des Tumors und dessen Behandlung; ermöglichen andererseits aber auch die spezifische oder symptomatische Behandlung des PNS.
PNS können unabhängig vom Tumorleiden zu schweren Behinderungen führen. Als Beispiel sind die PCD und die SSN zu nennen. Hypothetisch wird angenommen, dass eine frühe Therapie die Ausprägung des Vollbildes und der Behinderung aufhalten kann.
Diese Zusammenfassung weist auf klinische Symptome und Zeichen bei PNS hin. Besonders bei den PNS ist die klinische Beurteilung der wichtigste Baustein zum Nachweis der PNS und auch zur Entdeckung eines Tumorleidens.
1 Raspotnig M, Totland C, Storstein A, Vedeler C. What a Clinician Must Know Regarding Diagnosis of Paraneoplastic Neurological Syndromes. EANO Mag 2012; 2(2):67–70
2 Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol 2010; 257:509–17
3 Bien CG, Vincent A, Barnett MH et al., Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012; 135(Pt 5):1622–38.
4 Pelosof LC, Gerber DE. Paraneoplastic Syndromes: An Approach to Diagnosis and Treatment. Mayo Clin Proc 2010; 85:838–85
5 Titulaer MJ, Soffietti R, Dalmau J et al., European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol 2011; 18:19–e3
6 Giometto B, Grisold W, Vitaliani R et al., PNS Euronetwork. Paraneoplastic neurologic syndrome in the PNS Euronetwork database: a European study from 20 centers. Arch Neurol 2010; 67:330–5
7 Graus F, Delattre JY, Antoine JC et al., Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004; 75:1135–40
8 Zuliani L, Graus F, Giometto B et al., Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83(6):638–45
9 DeAngelis L, Posner JB. Neurological complications of cancer. 2nd edition Oxford University Press 2009; 580
10 Saiz A, Bruna J, Stourac P et al., Anti-Hu-associated brainstem encephalitis. J Neurol Neurosurg Psychiatry 2009; 80(4):404–7
11 Vigliani MC, Honnorat J, Antoine JC et al., PNS EuroNetwork. Chorea and related movement disorders of paraneoplastic origin: the PNS EuroNetwork experience. J Neurol 2011; 258:2058–68
12 Briani C, Vitaliani R, Grisold W et al., PNS Euronetwork. Spectrum of paraneoplastic disease associated with lymphoma. Neurology 2011; 22(76):705–10
13 Bayat E, Kelly JJ. Neurologic Complications in Plasma Cell Dyscrasias. Handbook of Clinical Neurology 104 and 105. Aminoff MJ, Boller F, Swaab DF (eds.); 3rd series 2012, Neurooncology, part 2; 731–746,
14 Grisold W, Vass A. Neuromuscular complications. Handbook of Clinical Neurology 104 and 105.
Aminoff MJ, Boller F, Swaab DF (eds.); 3rd series 2012, Neurooncology, part 2; 781–804
15 Waletzko-Niederdräing E, Mezger J, Mestarp ME et al., Paraneoplastische Degeneration des unteren Motoneurons bei Bence-Jones-Plasmozytom. Onkologe 1999; 5:734–737
16 Calabek B, Lindner K, Grisold W. Paraneoplastic Encephalitis, Myelitis, and Posterior Column Degeneration in a Patient with Breast Cancer. EANO Mag 2012; 2(2):93
17 Grisold W, Giometto B, Vitaliani, Oberndorfer S. Current approaches to the treatment of paraneoplastic encephalitis. Ther Adv Neurol Disord 2011; 4(4):237–248

AutorIn: Prim. Univ.-Prof. Dr.
Wolfgang Grisold
Neurologische Abteilung, Kaiser-Franz-Josef-Spital Wien

Ursprünglich erschienen:
neuro 04|2012
neuro 04|2012

