


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Die Identifikation neuer Targets in der CML
10. Juni 2011
Aus dieser Ernüchterung heraus sind Forschungsansätze hervorgegangen, die das Ziel verfolgen, weitere potenzielle Target-Moleküle in den involvierten Signalwegen, insbesondere in den leukämierepopulierenden Stammzellen zu identifizieren. Das dem BCR-ABLMolekül nachgeschaltete Signaling ist aufgrund der Vielzahl von beeinflussten Komponenten, deren Redundanz und Interaktion bisher noch wenig verstanden wird.
Es ist daher von zentraler Bedeutung, mit innovativen Hochdurchsatz-Methoden nach neuen Zielstrukturen zu suchen. Hierzu eignen sich insbesondere Technologien, die geeignete Ziele in den entsprechenden Zielzellen (wie z. B. den CML-repopulierenden Stammzellen) identifizieren lassen. Vor diesem Hintergrund wurden in den letzten Jahren Microarray- Studien durchgeführt: Mit Hilfe dieser Technologie lässt sich das mRNAProfil der entsprechenden Zellen analysieren und beispielsweise mit dem Profil gesunder hämatopoetischer Stammzellen vergleichen, bzw. können während der Krankheitsprogression oder unter TKI-Behandlung veränderte Signalwege identifiziert werden. Diese Studien wurden zudem initiiert, um über definierte genetische Profile bei der Erstdiagnose noch vor Therapiebeginn das Therapieansprechen vorauszusagen. Als Alternative zur Analyse von mRNA-Profilen dient die Analyse von Proteinexpressionsmustern mittels „Proteomics“ (z. B. 2-D-Gelelektrophorese, Massenspektrometrie), aber auch die Analyse von „Small Nuclear Polymorphisms“ (SNP) und von epigenetischen Genregulationsmustern (Methylierung) zur Target-Identifikation. Entsprechende Arbeiten als Grundlage für neuartige Therapieansätze sind für die CML bislang aber nur sehr limitiert verfügbar.
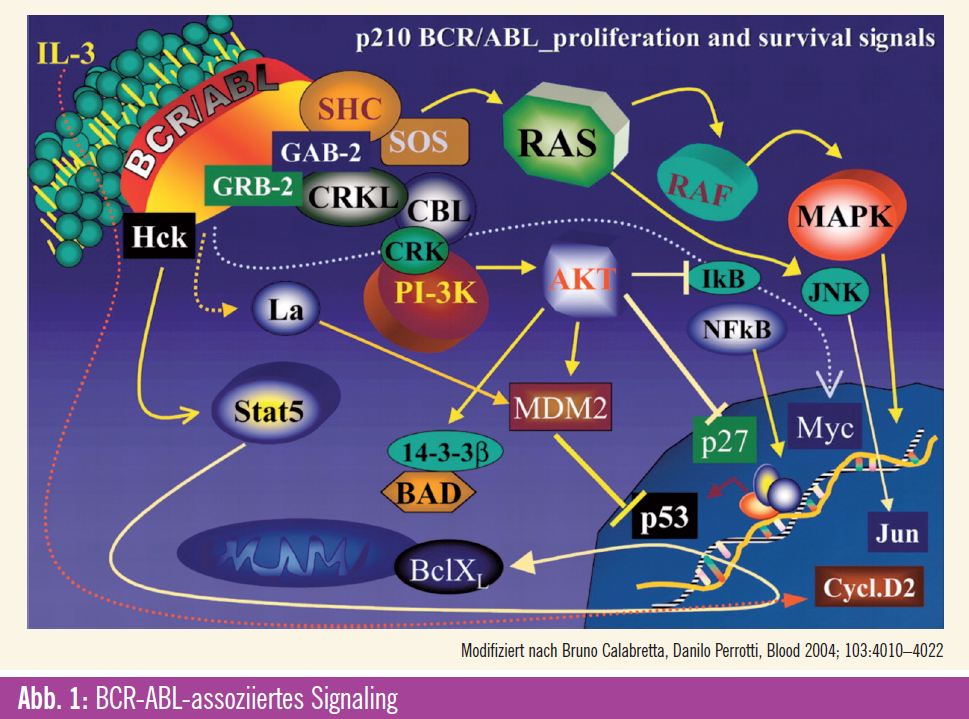
BCR-ABL-assoziiertes Signaling
Im Folgenden werden einzelne wichtige Signalwege besprochen, die unter anderem mittels oben genannter Technologien identifiziert werden konnten und als mögliche Ziele einer pharmakologischen Blockade potenziell die Krankheitsprogression limitieren könnten. Basis für eine solche Entwicklung stellt die Beobachtung dar, dass die Progression in die Blastenkrise der CML einerseits den Verlust der in der chronischen Phase noch erhaltenen Differenzierungsfähigkeit und andererseits die zunehmende Proliferationsrate darstellt. Weiters werden durch das BCR-ABL-Fusionsprotein zahlreiche Überlebens- und Apoptose-Signale dereguliert. Es konnte gezeigt werden, dass solche Effekte unter anderem auf einer Modulation der MAP-Kinase- und der PI3-Kinase-Kaskade beruhen1 (Abb. 1). Die Effektor-Kinase ERK vermittelt hierbei durch die Inhibition eines autokrinen extrinsischen Apoptose-Signalings einen antiapoptischen Effekt.2 In der CML ist bekannt, dass die ERK-Aktivität durch CCN3 supprimiert wird, das wiederum durch BCR-ABL induziert wird. Des Weiteren ist bekannt, dass MAP-Kinasen auch direkt Survival-Signale des BCL-2- Rheostat, wie BIM und MCL-1, modulieren. 3, 4 Diese Effekte sind jedoch nicht so ausgeprägt, dass eine alleinige Inhibition der MAP-Kinasen einen ausgeprägten antileukämischen Effekt induziert. Einen gegenteiligen Effekt hat hier das über p38 (eine weitere MAP-Effektor-Kinase) vermittelte Signaling, welches die Proliferation hemmt und gleichzeitig Apoptose begünstigt. Es konnte gezeigt werden, dass p38 durch Imatinib aktiviert werden kann, was eventuell mit zur apoptoseinduzierenden Wirkung von Imatinib beiträgt.
Für JNK2 (eine weitere MAP-Kinase) werden pro- wie auch antileukämische Effekte beschrieben. Ein proapoptotischer Effekt wird durch die BCR-ABL-vermittelte JNK2-Suppression unterdrückt1. Andererseits scheint JNK2 eine Rolle bei der CML-Transformation zu spielen und außerdem Autophagie (ein Prozess, bei dem ähnlich der Phagozytose Bestandteile einer Zelle abgebaut werden, wobei dies jedoch nur innerhalb einer Zelle anfallende Bestandteile betrifft) zu induzieren. 5, 6 Die Reduktion der Ribosomenzahl während der Autophagie vermindert auch die Expression von Tumorsuppressorgenen und verhindert so die Induktion von Apoptose. Im CML-Zelllinien-Modell (K562-Zellen) kommt es bei simultaner Behandlung mit Imatinib und einem Autophagie- Inhibitor zur vermehrten Apoptose.
BCR-ABL-Signaling beeinflusst darüber hinaus auch durch mTOR vermittelte Effekte auf das Zellwachstum, die Proliferation und die Regulation von Überlebenssignalen. Diese Wirkung wurde vor allem durch die Wirksamkeit von Rapamycin auf CML-Zellen nachgewiesen. Unter anderem mediiert mTOR solche Effekte durch die Regulation der Ubiquitinierung von PDCD4, das selbst ein Regulator der Translation von mRNA ist und als Tumorsuppressor fungiert. Durch das BCR-ABLSignaling wird die Expression von PDCD4 downreguliert und hebt damit dessen Wirkung als Tumorsuppressor auf.7
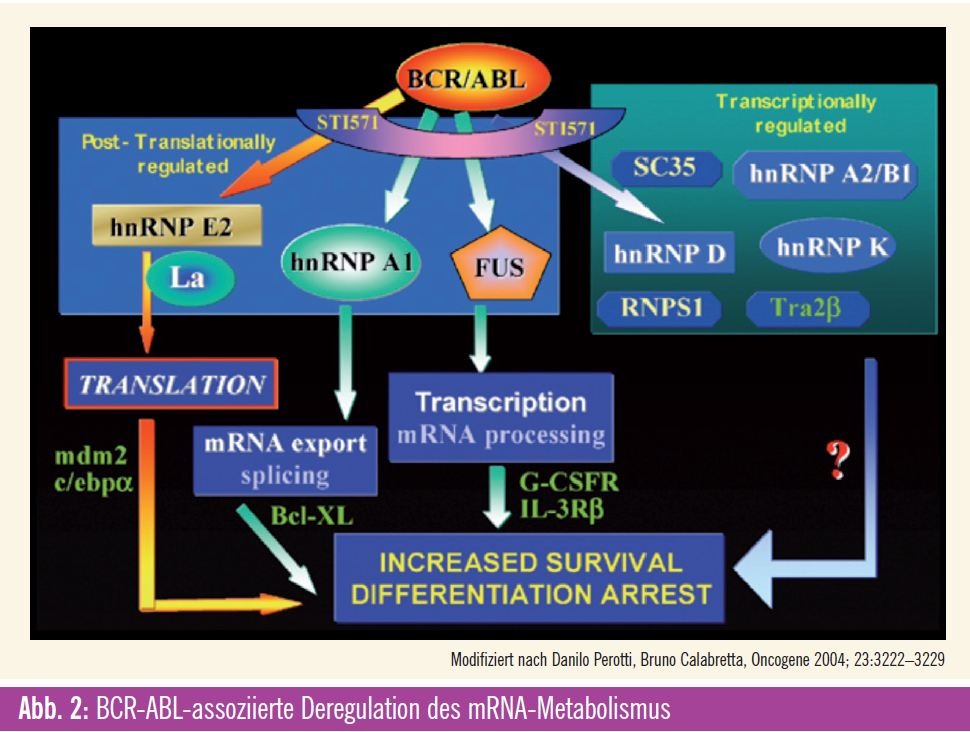
Deregulierte mRNA-Prozessierung
Neben klassischen Signalkaskaden, die über die gesteigerte Kinaseaktivität zur Aktivierung oder Inhibition von Schlüsselproteinen führen, oder der Regulation der Proteinstabilität wie im Beispiel von PDCD4 spielt auch die Modulation der Prozessierung von mRNA eine Rolle in der Progression der CML (Abb. 2). Beispielsweise kann alternatives Splicing zur atypischen Differenzierung führen. Hierbei sind Targets wie IKAROS8 involviert. Alternativ kann die Aktivierung des für die Selbsterneuerung von Stammzellen relevanten Wnt-Signaling durch eine Splice-Variante der GSK3beta induziert werden. Auch der nächste Schritt in der Prozessierung von mRNA kann durch Deregulation die Progression der CML fördern: Eines der den Zellzyklus positiv steuernden Cycline (Cyclin D3) wird BCR-ABL-abhängig vermehrt umgeschrieben (translatiert). Auf demselben Mechanismus basiert auch die Stimulation des Wachstumsfaktors VEGF, der durch eine vermehrte posttranskriptionelle Expression von HIF gesteuert wird. Eine Reihe von Proteinen kann die Prozessierung von mRNA auch sequenz – spezifisch steuern. Eine entsprechende Übersichtsarbeit von Perrotti et al. beschreibt die Bedeutung dieser Effekte in der CML.9 So führen hnRNP K und La/SSB zu einer vermehrten Translation unter anderem von MYC, MCL-1 und MDM2 (das die Ubiquitinierung von p53 induziert) und damit zur vermehrten Proliferation und einem gesteigerten Überleben in CML-BC-Zellen. Über den vermehrten nukleären Export von SET, BCL-XL und E2F3 werden vergleichbare Effekte zum Beispiel durch eine Überexpression von Cyclin E und eine Suppression des Tumorsuppressors PP2A erzielt. Die RNA-bindenden Proteine hnRNP E2, CUGBP1 hemmen die Translation von C/EBP-alpha und bewirken so einen Differenzierungsstopp. Dabei wird die Expression von hnRNP K und hnRNP E2 über die MAP-Kinase reguliert. Denselben Effekt erzielt eine durch den PI3-Kinase- Pathway vermittelte vermehrte Expression von TLS/FUS, das durch eine Modifikation der mRNA-Prozessierung des G-CSF-Rezeptors zu dessen Suppression führt.
Das als Kontrazeptivum eingesetzte Ormeloxifen zeigt in einem CML-Zelllinienmodell einen antileukämische Effekt, dem zwei Mechanismen zugrunde liegen: einerseits eine MAP-Kinase-abhängige Induktion der intrinsischen Apoptose und andererseits ein G0-G1-Zellzyklus – arrest, der über MBP1, einen negativen Regulator von MYC, vermittelt wird.10
Fazit
Aus der Komplexität der verschiedenen Signalwege lässt sich erahnen, dass die bisher verfügbaren Therapien – obwohl sie die zentrale Target-Kinase BCR-ABL sehr potent inhibieren – nicht in der Lage sind, die Erkrankung komplett zu eradizieren. Die klinische Erfahrung, dass ein Großteil der Patienten nach Absetzen des TKI einen molekularen Rückfall erleidet, bestätigt diese Annahme. Daraus ergibt sich, dass über moderne Technologien neuartige Zielstrukturen vor allem in CML-repopulierenden Stammzellen identifiziert werden müssen, die es ermöglichen, den Überlebensnerv dieser Zellen zu treffen und damit eine dauerhafte Elimination der Erkrankung erlauben. Es müssen daher in Zukunft mit großer Wahrscheinlichkeit Kombinationstherapien getestet werden, welche sich das Wissen aus den Target-Screens zunutze machen. Es bleibt die Hoffnung, dass dadurch nach dem Quantensprung in der CML-Therapie des letzten Jahrzehnts (der Einführung der TKI) der nächste große Schritt nach vorne gelingt, nämlich die Elimination resistenter CML-Zellen und CMLrepopulierender Stammzellen, um letztendlich eine dauerhafte Heilung zu erreichen.
1 Redig AJ, Vakana E, Platanias LC, Regulation of mammalian target of rapamycin and mitogen activated protein kinase pathways by BCR-ABL. Leuk Lymphoma; 52 Suppl 1:45–53
2 Chen KC, Liu WH, Chang LS, Suppression of ERK signaling evokes autocrine Fas-mediated death in arachidonic acid-treated human chronic myeloid leukemia K562 cells. J Cell Physiol; 222:625–34
3 Aichberger KJ, Mayerhofer M, Krauth MT et al., Low-level expression of proapoptotic Bcl-2-interacting mediator in leukemic cells in patients with chronic myeloid leukemia: role of BCR/ABL, characterization of underlying signaling pathways, and reexpression by novel pharmacologic compounds. Cancer Res 2005; 65:9436–44
4 Aichberger KJ, Mayerhofer M, Krauth MT et al., Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 2005; 105:3303–11
5 Puissant A, Robert G, Auberger P, Targeting autophagy to fight hematopoietic malignancies. Cell Cycle; 9:3470–8
6 Puissant A, Robert G, Fenouille N et al., Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res; 70:1042–52
7 Carayol N, Katsoulidis E, Sassano A, Altman JK, Druker BJ, Platanias LC, Suppression of programmed cell death 4 (PDCD4) protein expression by BCR-ABL-regulated engagement of the mTOR/p70 S6 kinase pathway. J Biol Chem 2008; 283:8601–10
8 Klein F, Feldhahn N, Herzog S et al., BCR-ABL1 induces aberrant splicing of IKAROS and lineage infidelity in pre-B lymphoblastic leukemia cells. Oncogene 2006; 25:1118–24
9 Perrotti D, Harb JG, BCR-ABL1 kinase-dependent alteration of mRNA metabolism: potential alternatives for therapeutic intervention. Leuk Lymphoma; 52 Suppl 1:30–44
10 Pal P, Kanaujiya JK, Lochab S et al., 2-D gel electrophoresis-based proteomic analysis reveals that ormeloxifen induces G0-G1 growth arrest and ERK-mediated apoptosis in chronic myeloid leukemia cells K562. Proteomics.

Ursprünglich erschienen:
SO 02|2011
SO 02|2011