


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Wirkung von Imiquimod in der Tumortherapie
19. Oktober 2012
Kontext
Imiquimod (R-837, S-26308) gehört zur Familie der Imidazoquinoline, Nukleosidanaloga, die Mitte der 1980er Jahre mit dem Ziel der Entwicklung antiviraler Substanzen synthetisiert wurden. Bereits vor mehr als einen Jahrzehnt wurde das therapeutische Potenzial von Imiquimod, einem synthetischen Immunmodulator, erkannt. 1997 wurde Imiquimod erstmals zur Behandlung von Condylomata acuminata (äußerliche Feigenwarzen) und 2004 für die lokale Therapie bestimmter Neoplasien wie die aktinische Keratose oder das superfizielle Basalzellkarzinom zugelassen1. Imiquimod wird für diese Zwecke als Creme (Aldara, enthält 5 % Imiquimod) eingesetzt. Kontrollierte klinische Studien zeigten, dass die äußerliche Applikation von Imiquimod zu lokalen Entzündungsreaktionen führt, die in den meisten Fällen von Erythemen und Erosionen begleitet werden. Dabei wirkt Imiquimod als Immunmodulator, der sowohl das angeborene als auch das erworbene Immunsystem aktiviert1. Verschiedene Zytokine und Chemokine, die das Immunsystem modulieren und zur topischen Rekrutierung von Immunzellen führen, werden in Zellen der Haut synthetisiert. Studien belegen, dass diese lokalen Hautreizungen eindeutig mit dem Therapieerfolg von Imiquimod in Verbindung stehen.
Wirkmechanismus
Imiquimod scheint seine Wirkung einerseits mittels verschiedenster Signaltransduktionswege zu entfalten. Unter anderem wurden die Inhibierung des Adenosin-Rezeptors A2a sowie die Induktion von Opioidrezeptoren (OGFr) beschrieben, die zu Apoptose bzw. verminderter Proliferation und damit in beiden Fällen zu reduziertem Tumorzellwachstum führte2, 3. Von diesen, vornehmlich an Tumorzelllinien gezeigten Effekten können andererseits die durch die agonistische Wirkung von Imiquimod an den Toll-like-Rezeptoren (TLR) 7 und 8 induzierten Mechanismen unterschieden werden4. TLR sind Erkennungsrezeptoren vor allem des angeborenen Immunsystems, die erregerassoziierte molekulare Muster erkennen und daher vorwiegend auf mikrobielle Komponenten reagieren. Verschiedene TLR findet man auf unterschiedlichen Zellen des angeborenen und erworbenen Immunsystems, wie zum Beispiel dendritischen Zellen (DC), B-Lymphozyten und Makrophagen, welche für die Präsentation von Antigenen und die Aktivierung des adaptiven Immunsystems wichtig sind. TLR 7 und 8 im Speziellen sind für die Erkennung von viraler Einzelstrang-RNA wie z. B. von Influenzaviren und HIV verantwortlich und befinden sich deshalb auch intrazellulär in den Endosomen, das sind Vesikel, die am Transport von Substanzen von der Membran in die Zellen beteiligt sind5.
Dendritische Zellen
DC sind antigenpräsentierende Zellen an der Schnittstelle zwischen angeborenem und erworbenem Immunsystem. Mit einer Vielzahl von Rezeptoren an ihrer Oberfläche und im Zellinneren sind sie spezialisiert für die Aufnahme und Erkennung von Erregern sowie Zelldebris6. Deren Abbauprodukte werden in Peptid-MHC-Komplexen an T-Zellen präsentiert, wodurch in Abhängigkeit von Stimulus und Zytokinmilieu eine antigenspezifische Immunantwort initiiert oder Toleranz induziert werden kann. Andererseits können Zytokine wie z. B. von plasmazytoiden DC (pDC) produziertes Typ-I-Interferon auch Zellen des angeborenen Immunsystems aktivieren. Innerhalb der Familie der DC werden TLR7 und 8 in Menschen von pDC und myeloiden DC (mDC), in Mäusen von pDC und CD4+ mDC exprimiert7. Die Aktivierung durch TLR7/8-Agonisten wie Imiquimod führt in den DC über das Adapterprotein MyD88 zur Aktivierung von Nf-κB und MAP-Kinasen und damit zur Produktion und Ausschüttung verschiedener Zytokine wie Interferon (IFN)-α und β, INF-γ, Interleukin (IL-) 6, 8, 12, 23, Tumornekrosefaktor (TNF)-α, G-CSF oder GM-CSF8. Weiters wird durch TLR7/8-Stimulation die Reifung der DC induziert, wodurch Faktoren wie CD80, CD86, CD40 und CCR7 vermehrt an der Zelloberfläche exprimiert und somit eine gesteigerte antigenspezifische T-Zell-Immunantwort induziert wird9, 10. Gemeinsam mit dem Zytokinprofil führt dies zur Differenzierung von naiven T-Zellen zu T-Helfer- (Th)-1 und Th-17-Zellen, die bei antiviralen, Antitumor-, aber auch bei Autoimmunreaktionen eine entscheidende Rolle spielen11, 12.
Plasmazytoide dendritische Zellen: Topische Behandlung mit Imiquimod bewirkt einerseits die Emigration von Langerhans-Zellen aus der Epidermis in die regionalen Lymphknoten, andererseits werden pDC wahrscheinlich direkt aus dem Blut in die Haut oder in den Tumor rekrutiert13–15. Die Migration und Reifung der Langerhans-Zellen in die regionalen Lymphknoten führt vermutlich zu einer vermehrten T-Zell-Stimulierung. Tatsächlich werden in Imiquimod-behandelten Basalzellkarzinomen vermehrt aktivierte T-Zellen, vor allem CD4+-T-Zellen, gefunden16. Ein definitiver Beweis für einen solchen Mechanismus konnte jedoch bis jetzt nicht erbracht werden. Die in den Tumor einwandernden pDC scheinen vor allem über ihre IFN-α/β-Produktion zu wirken. Die Faktoren, welche bei der Einwanderung der pDC eine entscheidende Rolle spielen, sind noch nicht genau bekannt. In der Haut residierende Mastzellen exprimieren auch TLR7. Im Mausmodell wurde gezeigt, dass das Fehlen von Mastzellen oder ein Defekt in der Produktion von Tumornekrosefaktor-α die Entzündung in der Haut sowie das pDC-Infiltrat verhindern17. Imiquimod-Behandlung führt zur Produktion einer ganzen Reihe von Chemokinen in der Haut, die für die Rekrutierung der pDC verantwortlich sein können. Unter anderem konnte in Tiermodellen gezeigt werden, dass in der Abwesenheit eines funktionierenden CCR6 und CCR10 oder CCL2 nicht vermehrt pDC nach Stimulation mit Imiquimod in die Haut einwandern18, 19. Welche Zellen in der Haut die Liganden, CCL20, CCL27 oder CCL2, sezernieren, muss jedoch erst genau bestimmt werden.
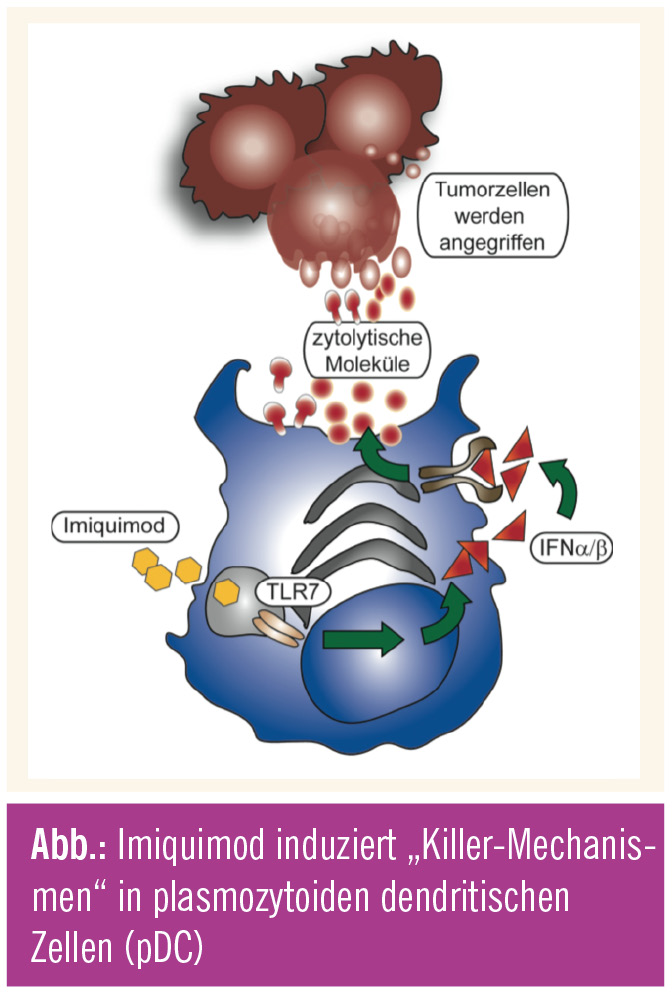
Toll-like-Rezeptoren, Interferon: Durch TLR7 aktivierte pDC sezernieren in vitro aber auch in vivo vor allem Typ-I-Interferone (IFN-α/β) und sind damit die wichtigsten IFN-α-produzierenden Zellen des Immunsystems. INF-α hat antivirale und antitumorigene Wirkung und ist einer der effizientesten Faktoren für die Aktivierung von mDC, natürlichen Killerzellen und T-Zellen, wodurch TLR-stimulierte pDC eine Schlüsselrolle im Immunsystem bei der Induktion und Modulation von Immunantworten übernehmen. Neben den Aufgaben als IFN-α-produzierende und antigenpräsentierende Zellen wurde vor Kurzem eine neue Funktion der Imiquimod-aktivierten pDC entdeckt, jene als Killer von Tumorzellen. Humane und murine Studien zeigen, dass mit Imiquimod stimulierte pDC nicht nur vermehrt T-Zell-stimulierende, kostimulatorische Moleküle exprimieren, sondern auch die zytolytischen Moleküle TRAIL (TNF-verwandter, Apoptose induzierender Ligand) und Granzym B hochregulieren und mit diesen Tumorzellen angreifen können. Neben dem TLR7 spielt bei dieser Killerfunktion IFN-α die entscheidende Rolle. Über einen autokrinen Mechanismus (sezerniertes Typ-I-IFN wirkt auf die pDC selbst ein) werden pDC stimuliert, um zytolytische Moleküle zu produzieren und an die Umgebung abzugeben (Abb.)18, 20. Diese zytolytischen Moleküle attackieren die Tumorzellen und lösen in diesen eine Signalkaskade aus, die zu deren Zelltod führt18, 20. In vitro Studien konnten auch zeigen, dass Stimulation von mDC mit Imiquimod ebenfalls zur Produktion der zytolytischen Moleküle Perforin und Granzym B führt und diese damit zytolytische Eigenschaften erwerben21.
Vakzinierungsstrategien: TLR-Liganden werden schon seit Jahrzehnten als Adjuvantien zur Immunisierung verwendet. In den letzten Jahren versucht man zunehmend, diese Eigenschaften auch für Vakzinierungsstrategien gegen Tumoren einzusetzen22. Zu diesem Zweck werden patienteneigene DC entnommen, vermehrt, mit Tumorantigenen beladen und wieder in den Patienten injiziert. Neben der Frage, welche DC-Subpopulation am besten geeignet ist, stellen die Migration der DC sowie deren optimale Stimulation die größten Herausforderungen dar. Die TLR7-Stimulation mit Imiquimod hat hier vielversprechende Ergebnisse gezeigt. Topische Applikation von Imiquimod zeitgleich oder vor der Gabe des Antigens oder antigenbeladener DC führt zu verbesserter Reifung und Migration der DC und damit zu einer effizienteren Immunisierung von T- und B-Zellen bei gleichzeitig guter Verträglichkeit23–25. In Zukunft werden neue Strategien, in welchen spezifisch mehrere TLR gleichzeitig stimuliert werden, diesen Therapieansatz einer erfolgreichen klinischen Verwendung noch näher bringen.
1 Tyring A, AG, SK, Rosen T, J Drugs Dermatol, 2009; 8(5):467–74.
2 Schon MP, Schon M, Expert Opin Ther Targets, 2006; 10(1):69–76.
3 Zagon IS et al., Exp Biol Med (Maywood), 2008; 233(8):968–79.
4 Schon MP, Schon M, Br J Dermatol, 2007; 157 Suppl 2:8–13.
5 Kawai T, Akira S, Nat Immunol, 2010; 11(5):373–84.
6 Mellman I, Steinman RM, Cell, 2001; 106(3):255–8.
7 Edwards AD et al., Eur J Immunol, 2003; 33(4):827–33.
8 Gorden KB et al., J Immunol, 2005; 174(3):1259–68.
9 Larange A. et al., J Leukoc Biol, 2009; 85(4):673–83.
10 Dang Y et al., Clin Cancer Res, 2012; 18(11):3122–31.
11 Lombard V, Akbari O, Drug News Perspect, 2009; 22(8):445–51.
12 van der Fits L et al., J Immunol, 2009; 182(9):5836–45.
13 Palamara F et al., J Immunol, 2004; 173(5):3051–61.
14 Tripp CH et al., Immunobiology, 2008; 213(9–10):715–28.
15 Urosevic M et al., J Natl Cancer Inst, 2005; 97(15):1143–53.
16 De Giorgi V et al., Int J Dermatol, 2009; 48(3):312–21.
17 Heib V et al., Blood, 2007; 110(3):946–53.
18 Drobits B et al., J Clin Invest, 2012; 122(2):575–85.
19 Sisirak V et al., Blood, 2011; 118(19):5130–40.
20 Kalb ML et al., J Immunol, 2012; 188(4):1583-91.
21 Stary G. et al., J Exp Med, 2007; 204(6):1441–51.
22 Palucka K, Banchereau J, Nat Rev Cancer, 2012; 12(4):265–77.
23 Nair S. et al., J Immunol, 2003; 171(11):6275–82.
24 Adams S. et al., J Immunol, 2008; 181(1):776–84.
25 Flacher V. et al., J Immunol, 2012; 188(5):2146–55.

Ursprünglich erschienen:
SO 03|2012
SO 03|2012
Herausgeber: Univ.-Prof. Dr. Christoph Zielinski, Univ.-Prof. Dr. Markus Raderer
Publikationsdatum: 2012-10-19
Zur Ausgabe »
Publikationsdatum: 2012-10-19
Zur Ausgabe »
