


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Psychopharmakainteraktionen – ein Update
21. Dezember 2012
Neben den bekannten Risikofaktoren wie höheres Alter, Multimorbidität und der zwangsläufig daraus resultierenden Polypharmazie erhöht die häufig sequenzielle Umstellung auf andere Substanzen und Substanzkombinationen während der Therapie mit Psychopharmaka zusätzlich das Risiko für Arzneimittelwechselwirkungen.
Dass Arzneimittelinteraktionen und nachfolgende unerwünschte Arzneimittelwirkungen (UAW) Morbidität, Hospitalisierungsrate und auch Mortalität erhöhen, ist evident. Im extramuralen Bereich finden sich bei 9–70 % der Patienten Arzneimittelinteraktionen, von denen 1–23 % als schwer zu bezeichnen sind. Während eines Krankenhausaufenthaltes erhöht sich mit der Anzahl an Arzneimitteln auch die der Interaktionen. Daten aus einer prospektiven Studie an 18.820 Patienten, die an zwei englischen Krankenhäusern über einen Zeitraum von 17 Monaten lief, zeigen, dass bei 1.225 Patienten UAW der Grund für die Aufnahme war. 72 % der Aufnahmen wären vermeidbar gewesen, 17 % der UAW gingen auf Arzneimittelinteraktionen zurück.
Speziell die Beachtung pharmakokinetischer Interaktionen stellt aufgrund der verschiedenen Substanzkombinationen und des zunehmenden Wissens um den über verschiedene Enzyme- und Transportproteine ablaufenden Arzneistoffmetabolismus eine Herausforderung dar und erfordert gute Grundlagenkenntnisse. Neben dem Einsatz von Datenbanken ist die Zusammenarbeit mit klinischen Pharmazeuten eine wertvolle Unterstützung in der Beurteilung der klinischen Relevanz von Arzneimittelinteraktionen.
Pharmakokinetische Interaktionen Interaktionen können dem LADME-Prinzip folgend auf allen pharmakokinetischen Ebenen stattfinden: beginnend mit der Freisetzung des Arzneistoffs aus der Arzneiform (L = Liberation) über die Resorption (A = Adsorption), die Verteilung in das umliegende Gewebe (D = Distribution) und die Metabolisierung (M = Metabolism) bis zur Ausscheidung (E = Elimination). Besonders relevant sind die Arzneimittelinteraktionen auf der Ebene des Metabolismus.
Die für psychotrope Substanzen maßgeblichen Gewebe sind die Enterozyten, Hepatozyten und die Endothelien der Blut-Hirn-Schranke. Hier erfolgt die Aufnahme in die Zelle, die Metabolisierung und der Auswärtstransport in einem sehr komplexen Zusammenspiel aus Transportproteinen und metabolisierenden Enzymsystemen. Das für den Arzneistoffmetabolismus wichtigste und am besten erforschte Enzymsystem ist das Cytochrom-P450-System (siehe Fact-Box). Die inhibitorischen und induzierenden Effekte unterliegen einem Zeitfaktor. Während die hemmende Wirkung auf den Abbau eines Arzneistoffes sofort nach Zugabe eines Inhibitors einsetzt, tritt die induzierende Wirkung mit zeit licher Verzögerung von Tagen bis Wochen ein, sofern der den Abbau induzierende Arzneistoff neu eingesetzt wird. Auch nach Absetzen des Arzneistoffes kann der induzierende Effekt noch 2–3 Wochen anhalten.
Psychopharmaka werden intensiv über verschiedene CYP-Isoenzyme verstoffwechselt. Dabei spielen hauptsächlich die Aktivitäten der Isoenzyme CYP3A, CYP2D6 und CYP1A2 eine Rolle.
CYP1A2: Clozapin wird hauptsächlich über CYP1A2 und im Nebenweg über CYP3A4 abgebaut. In Komedikation mit starken CYP1A2- und CYP3A4-Inhibitoren steigt der Clozapinspiegel an und damit die Wahrscheinlichkeit von UAW (Leukopenie, Neutropenie, EPMS, vermehrter Speichelfluss etc.). Schwere respiratorische Infekte können ebenfalls zu stark erhöhten Plasmaspiegeln mit teilweise deliranten Zustandsbildern führen. Als Ursache wird eine durch die Inflammation erniedrigte Aktivität von CYP1A2 angenommen. Im Gegenzug dazu beschleunigen potente CYP1A2- und CYP3A-Induktoren den Abbau von Clozapin und führen zu unter Umständen stark erniedrigten Clozapinspiegeln bzw. dessen Wirkverlust (Tab. 1).
Die Kombination von ebenfalls im Hauptweg über CYP1A2 verstoffwechseltem Agomelatin mit starken CYP1A2-Hemmern ist kontraindiziert (12–412-facher Anstieg des Agomelatinspiegels).
Das mit Clozapin chemisch verwandte Olanzapin benützt zum Teil die gleichen Abbauwege wie Clozapin, weist aber ein geringeres Risiko für „drug-drug interactions“ auf, da vor allem über die Glukuronidierung ein alternativer Abbauweg zur Verfügung steht. Aufgrund der im Alter eingeschränkten Aktivität des CYP450-Systems muss die Dosis entsprechend angepasst werden. Rauchen führt sowohl bei Clozapin wie auch bei Olanzapin, Agomelatin und Duloxetin zu erniedrigten Plasmaspiegeln und damit zu Wirkverlust (Tab. 1).
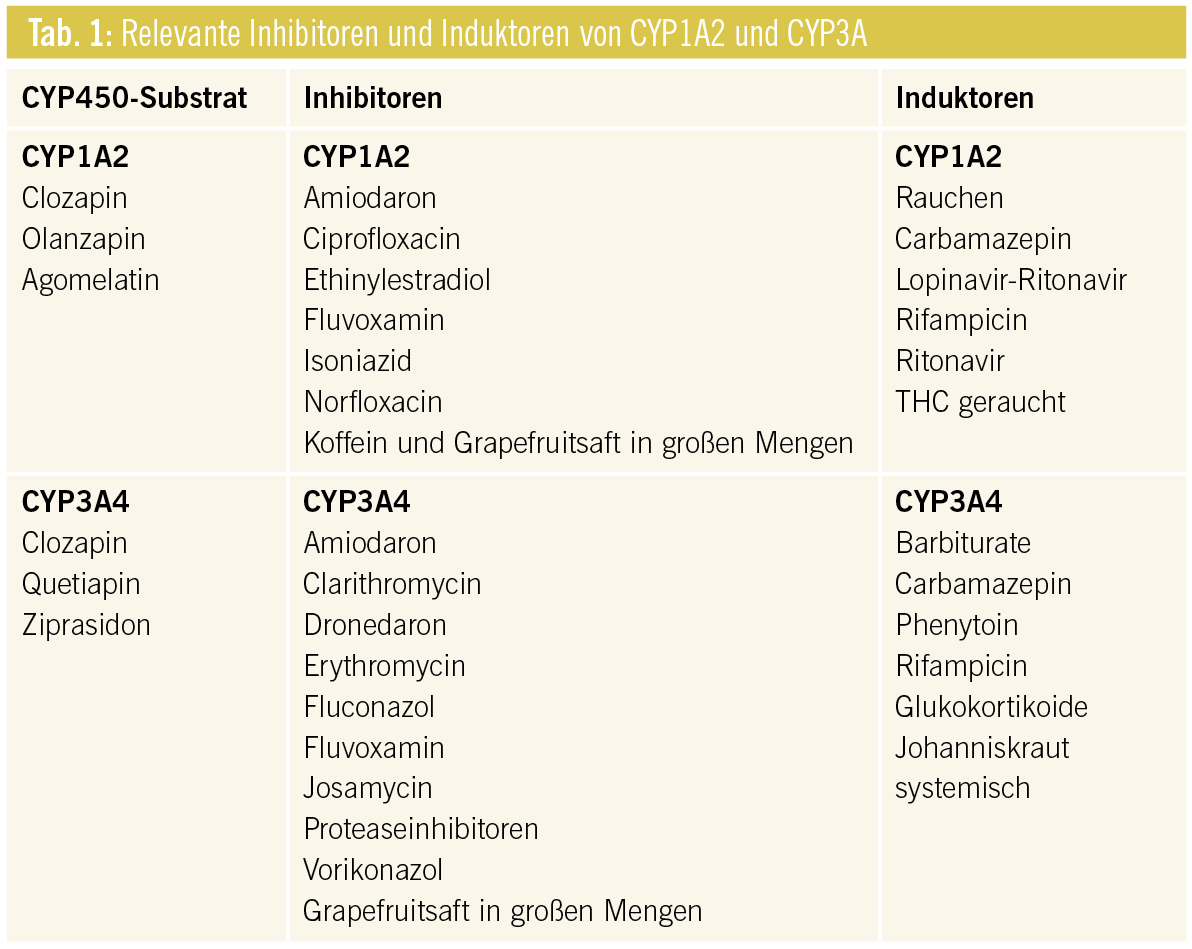
CYP2D6: Für den Abbau von Aripiprazol und Risperidon spielt CYP2D6 ebenso eine Schlüsselrolle wie für die meisten Antidepressiva der neuen Generation. Fluoxetin, Paroxetin und Sertralin in hoher Dosierung sind nicht nur Substrate von CYP2D6, sondern hemmen dieses auch wie Haloperidol und Bupropion (Tab. 2). Bei rezidivierenden Depressionen können Duloxetin und Venlafaxin in Kombination mit dem CYP2D6-Hemmer Bupropion wegen der pharmakokinetischen und dynamischen Wirkverstärkung vermutlich zur Augmentationstherapie eingesetzt werden.
Bei Kombination von Risperidon mit den SSRI Paroxetin, Fluoxetin und Sertralin in Hochdosis resultieren erhöhte Plasmaspiegel und verstärkte UAW (EPMS, QTc-Verlängerung, Zunahme des Prolaktinspiegels, Orthostasen etc.).
Paliperidon (9-Hydroxy-Risperidon), der mit einer HWZ von 24 h lang wirksame, aktive Hauptmetabolit von Risperidon, kann wegen seines geringen Wechselwirkungspotenzials (nur Phase-II-Metabolisierung) mit Vorteil bei Leberinsuffizienz und bei Polypharmazie eingesetzt werden. Allerdings erfordern Niereninsuffizienz und höheres Alter eine Dosisanpassung, da 60 % unverändert renal ausgeschieden werden.
Ein weiteres Antipsychotikum mit minimalem Potenzial für Interaktionen über CYP450 ist Amisulprid, das wie Paliperidon zu 50 % unverändert über die Niere ausgeschieden wird. Eine Dosisanpassung bei eingeschränkter Leberfunktion ist auch hier nicht notwendig, bei Niereninsuffizienz sehr wohl erforderlich.
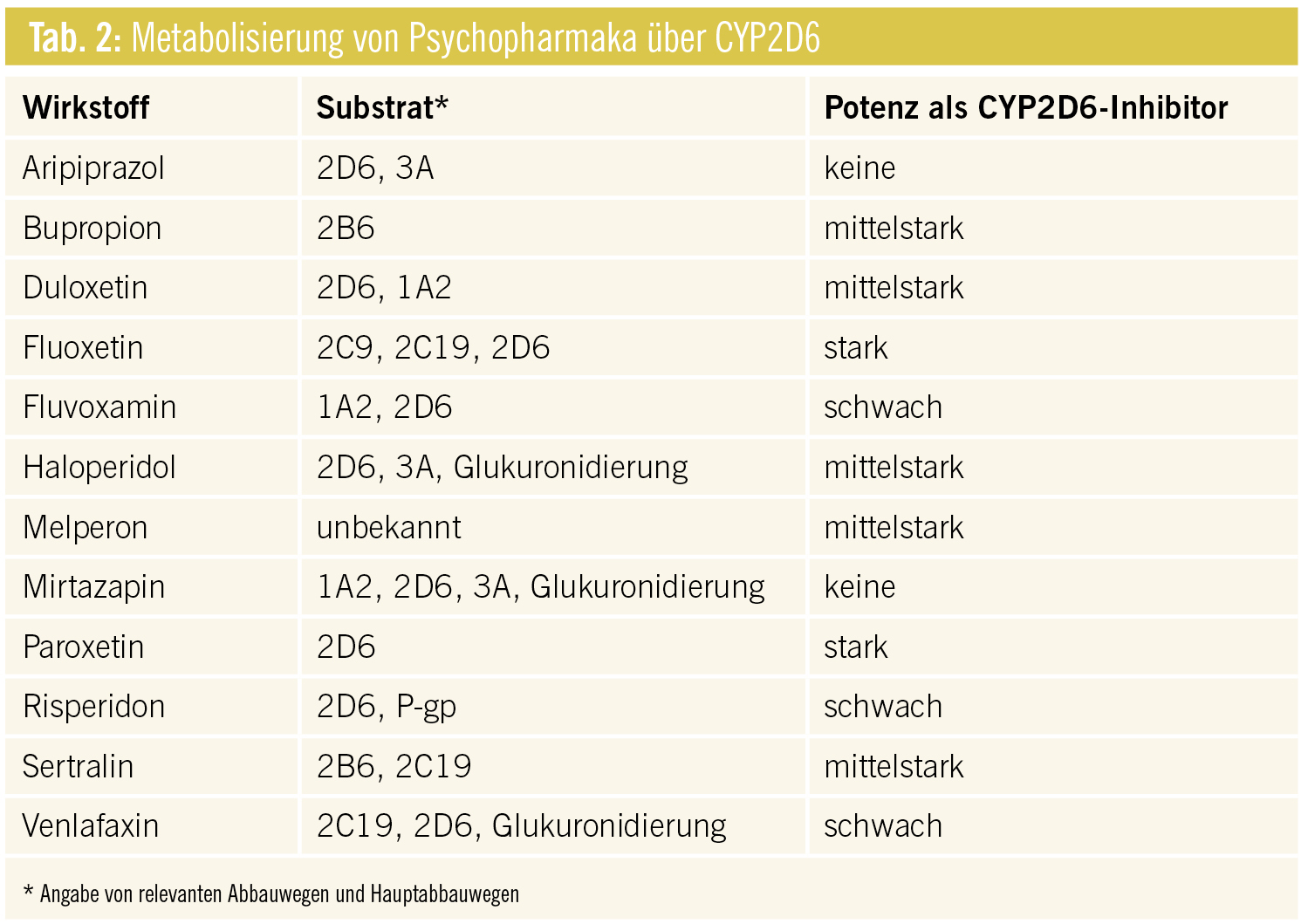
CYP3A4: Quetiapin und Ziprasidon werden beinahe exklusiv über CYP3A4 verstoffwechselt. Starke CYP3A4-Inhibioren lassen Plasmaspiegel bzw. die Rate an UAW ansteigen (Sedierung, Orthostase, Verlängerung des QT-Intervalls etc.). Ältere Patienten reagieren besonders rasch mit Übersedierung und orthostatischer Hypotonie und erhöhen damit ihr Sturzrisiko. Im Gegenzug führt die Komedikation mit potenten CYP3A4-Induktoren zu beschleunigtem Abbau und Wirkverlust der genannten Substanzen (Tab. 1).
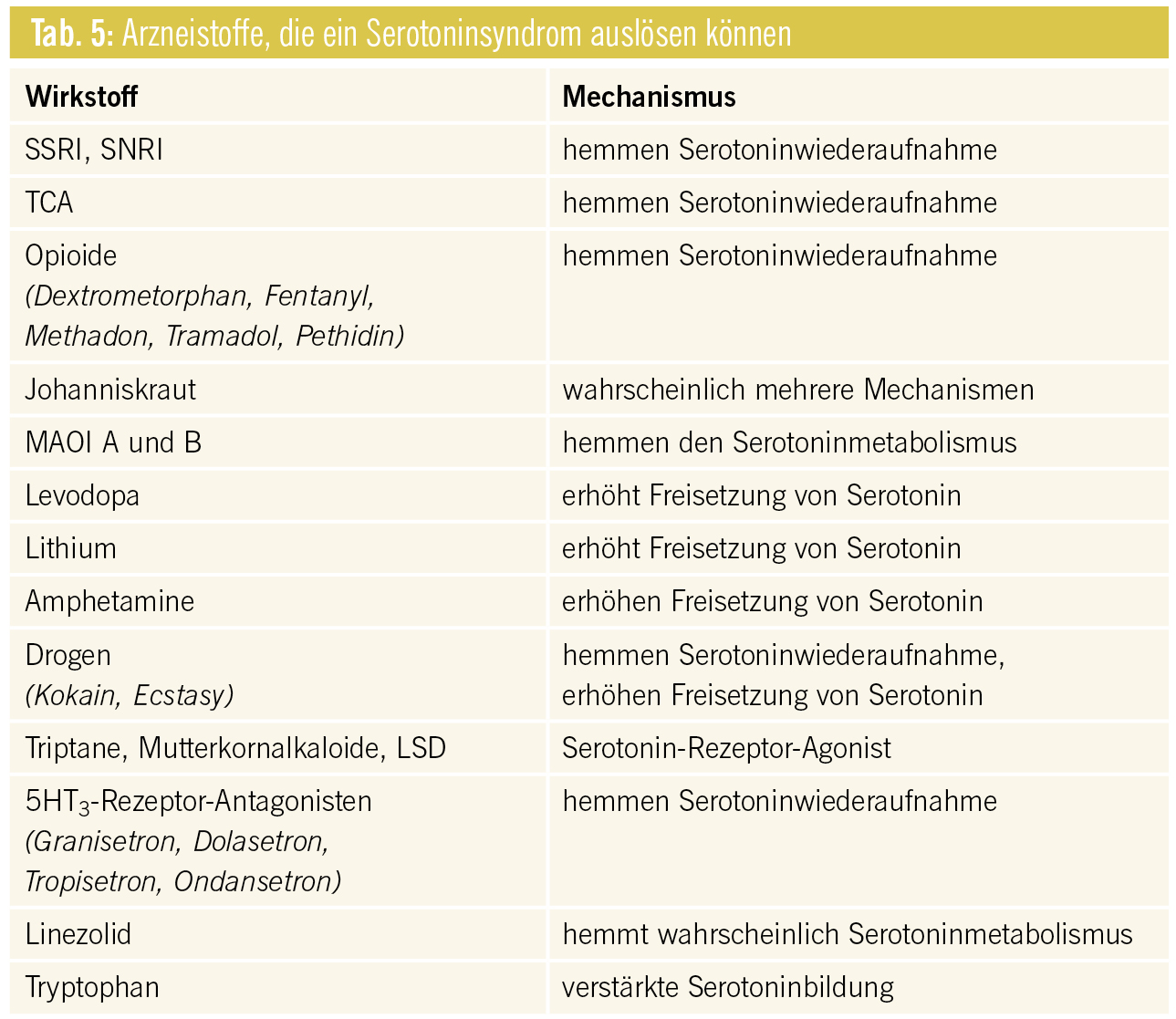
Pharmakodynamische Interaktionen Additive serotonerge Wirkungen: In der Regel kann die Kombination von zwei oder mehr Substanzen mit serotonergem Wirkmechanismus (v. a. MAO-Hemmer, SSRI, SNRI, Antipsychotika vom Phenothiazintyp), Dosiserhöhung oder die Kombination mit einem abbauhemmenden Arzneistoff zu erhöhter Serotonintoxizität führen. Durch eine Überstimulierung der postsynaptischen 5-HT1A und 5-HT2-Rezeptoren können sich innerhalb von 24 Stunden unerwünschte zentrale und periphere Wirkungen ergeben. Die Sensitivität auf die verstärkte Serotoninwirkung bzw. die Ausprägung der klinischen Symptomatik ist individuell verschieden. Sie reicht von Tremor, Unruhe- oder Angstzuständen bis zu tonisch- klonischen Krämpfen und Delir (Tab. 3).

Die Diagnostik des in der klinischen Praxis selten vorkommenden Serotoninsyndroms kann nach den Sternbach- oder Hunter-Kriterien erfolgen, wobei Letztere einfacher anzuwenden sein dürften (Tab. 4).

Die Therapie des Serotoninsyndroms liegt im Absetzen der auslösenden Substanzen. In leichteren Fällen klingt das Syndrom innerhalb von 24–72 Stunden ab, schwere Verläufe erfordern den Einsatz von Benzodiazepinen und intensivmedizinische Betreuung.
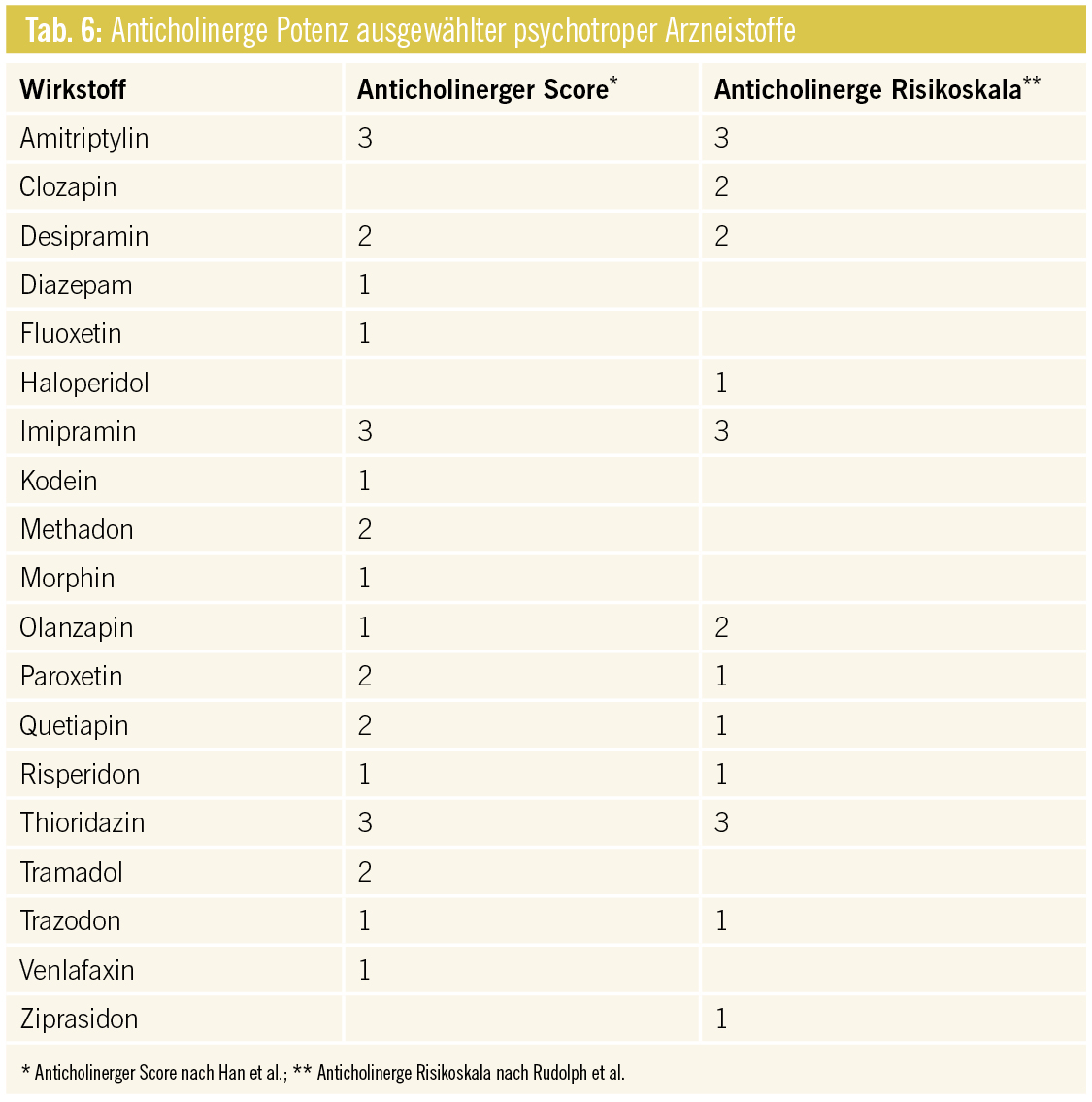
Additive anticholinerge Wirkungen: Diese können vor allem in der Therapie mit trizyklischen Antidepressiva, Antipsychotika vom Phenothiazintyp und manchen Benzodiazepinen auftreten. Das kumulative Risiko für zentrale und periphere anticholinerge Effekte erhöht sich bei Komedikation mit weiteren anticholinerg wirkenden Arzneistoffen. Anticholinerge Nebenwirkungen werden vom Patienten als besonders unangenehm empfunden und beeinflussen die Adhärenz negativ.
Besonders ältere Patienten sind potenziell häufiger davon betroffen, da die Azetylcholinsynthese durch Verarmung cholinerger Neurone im Alter abnimmt. Eine anticholinerge Medikation oder interaktionsbedingte additive Effekte können zu Gedächtnisstörungen und Verwirrtheit bis hin zum Delir führen. An der Peripherie äußern sich unerwünschte anticholinerge Arzneimittelwirkungen in Harnverhalt, Obstipation, Mundtrockenheit, Tachykardie und Akkommodationsstörungen am Auge.
In der klinischen Praxis eignet sich zur Bestimmung des kumulativen Risikos bzw. der anticholinergen Gesamtbelastung beim Patienten die Kenntnis der anticholinergen Potenz eines Arzneistoffes unter Berücksichtigung von Parametern wie Grunderkrankungen, Organinsuffizienzen, Alter und Dosis (Tab. 6). Häufig verordnete nichtpsychotrope Arzneistoffe mit erhöhtem anticholinergem Potenzial sind Urologika (v. a. Oxybutinin, Tolterodin), Antihistaminika (Diphenhydramin, Dimenhydrinat, Hydroxyzin), Gyrase-Hemmer, das Muskelrelaxans Orphenadrin und Antiparkinsonia (Amantadin, Biperiden, Dopaminantagonisten, L-Dopa in hoher Dosierung).
Additive kardiotoxische Wirkungen: Die Verlängerung des QT-Intervalls im Oberflächen-EKG ist eine in der Psychopharmakatherapie häufige, unerwünschte Arzneimittelwirkung. Lange Zeit unterschätzt, hat dieses Thema in den letzten Jahren zunehmend an Bedeutung gewonnen, da einige Arzneimittel mit nichtkardialer Indikation wegen dieser schwerwiegenden Nebenwirkung vom Markt genommen werden mussten. Durch Hemmung transmembranöser Kaliumströme kommt es zu einer überschießenden QT-Verlängerung, die zu lebensbedrohlichen ventrikulären Herzrhythmusstörungen vom Torsade-depointes- Typ führen kann. Da die QT-Dauer von der Herzfrequenz abhängig ist – bei Bradykardie nimmt sie zu, bei Tachykardie nimmt sie ab – wird als Vergleich die frequenzkorrigierte QT-Zeit herangezogen (Tab. 7).
Risikofaktoren für eine Verlängerung des QTc-Intervalls und die Entwicklung von Torsade de pointes sind: weibliches Geschlecht, Bradykardie, Hypokaliämie und deren Ursachen (Diuretika, Erbrechen, Diarrhö), schwere Hypomagnesiämie, Hypokalzämie, Anorexie, paroxysmales Vorhofflimmern, Herzinsuffizienz oder schwere Kardiomyopathie, Niereninsuffizienz, verlängertes QT-Intervall auf dem Basis-EKG, Komedikation von Medikamenten mit Potenzial für QT-Verlängerung, Abbauhemmung des Medikamentes mit QT-verlängernder Wirkung sowie angeborene QT-Verlängerung.
Antipsychotika wie Sertindol, Pimozid, Ziprasidon oder Haloperidol sowie die Antidepressiva Venlafaxin, Sertralin in Hochdosis oder Citalopram in Hochdosis besitzen ein teilweise hohes Potenzial für QT-Zeit-Verlängerung und Torsaden (Tab. 8).
Bei Verlängerung des QT-Intervalls um 45–60 ms bzw. bei absoluter Dauer von > 500 ms oder bei Auftreten von Torsaden muss die auslösende Medikation abgesetzt, der Patient eventuell überwacht und eine verträglichere Alternative gefunden werden (aktualisierte Informationen zu Arzneimitteln, die das QT-Intervall verlängern: www.torsades.org, www.azcert.org, www.sads.org).


Erhöhtes Blutungsrisiko unter SSRI/SnRI
Das in Thrombozyten enthaltene Serotonin fördert die Plättchenaggregation und damit die Hämostase, kann von den Blutplättchen aber selbst nicht gebildet werden. SSRI und auch SNRI führen zu einem Serotoninmangel im Thrombozyten, was zu verminderter Aggregation und verlängerter Blutungszeit führt. Die Datenlage der letzten Jahre, ob SSRI alleine oder in Kombination mit Antikoagulantien das Blutungsrisiko erhöhen, ist divergent.
Einerseits kommen frühere Studien und Reviews zu dem Schluss, dass SSRI das Risiko für GI-Blutungen speziell im oberen GI-Trakt erhöhen und sich dieses unter Gabe von ASS/NSAR potenziert. Andere Studien fanden kein erhöhtes Risiko für Blutungen, auch nicht im oberen GI-Trakt. Ähnliches gilt für die Kombination mit Phenprocoumon. Weiterhin gilt für SSRI und SNRI eine direkte Steigerung der Magensäureproduktion als unerwünschte Arzneimittelwirkung, wodurch ebenso wie durch den längerfristigen Gebrauch von NSAR die Bildung von Ulzera sowie GI-Blutungen begünstigt wird. Ein neuerer Review von Andrade et al. beschreibt ein moderat erhöhtes Risiko für eine GI-Blutung unter der Therapie mit SSRI. Im Einzelfall müssen entsprechend der Komorbidität (GI-Blutungen in der Anamnese, Langzeittherapie mit NSAR, hereditäre Blutungsanomalien) und dem Alter des Patienten der Einsatz einer Ulkusprophylaxe (HT2-Antagonisten, PPI in niedriger Dosierung) oder Medikamentenänderungen in Betracht gezogen werden.
Fazit
Um die klinische Relevanz einer möglichen, in diversen Interaktionstools beschriebenen Arzneimittelinteraktion zu beurteilen, müssen folgende Zusatzinformationen vorliegen:
- aktueller Blutbefund: Elektrolyte, Nierenfunktions- und Leberfunktionsparameter (zur Erfassung pharmakokinetischer Aspekte und des momentanen Istzustands des Patienten)
- aktuelle Diagnosen (zur Nutzen-Risiko-Bewertung des derzeitigen Arzneimitteleinsatzes)
- eventuell vorhandene Symptome, verstärkte Nebenwirkungen …
Unter Berücksichtigung dieser Aspekte ist es trotzdem oft schwierig, die klinische Relevanz einer möglichen, im Interaktionsprogramm mit „schwer“ angegebenen Interaktion im individuellen Fall zu bewerten. Kontraindikationen, d. h. Kombinationen, die laut Fachinformation nicht eingesetzt werden dürfen, müssen aber auf alle Fälle berücksichtigt werden. Ansonsten gilt oft ein „CAVE“: unter Beobachtung möglich, eventuell auf Arzneimittel mit höherem Sicherheitsprofil umstellen (unter Einbeziehung der oben beschriebenen Zusatzinformationen). Als Faustregel gilt: Je geringer die therapeutische Breite des Wirkstoffes und je größer die Organbeeinträchtigung beim Patienten sind, desto größer ist die Wahrscheinlichkeit von klinisch relevanten Wechselwirkungen.
Fact-Box
Metabolisierung von Arzneistoffen
Das Cytochrom-P450-system umfasst mehrere Familien und Subfamilien, die den oxidativen Abbau von Arzneistoffen in der Phase i des Metabolismus katalysieren. Arzneistoffe sind nicht nur Substrate eines oder mehrerer Isoenzyme, sie können deren Enzymfunktion auch hemmen (Inhibitoren) oder induzieren (Induktoren). in der Folge steigen bei bestimmten Arzneimittelkombinationen die Blutspiegel der betroffenen Arzneistoffe an, UAW treten verstärkt auf. im Gegenzug können die Blutspiegel der betroffenen Arzneistoffe absinken, woraus ein Wirkverlust resultiert. Über 50 % der Arzneistoffe werden über das quantitativ wichtigste Isoenzyme CYP3A abgebaut. Die Menge der einzelnen Isoenzyme ist intraindividuell sehr unterschiedlich, da sie genetischen Polymorphismen unterliegen.
Die UDP-Glukuronosyltransferasen (UGT) übertragen in Phase II des Metabolismus Glukuronsäure auf Substrate. die daraus resultierenden, hydrophileren Metabolite sind im Allgemeinen inaktiv, können aber in seltenen Fällen auch aktive Metaboliten oder potenter als die Muttersubstanz sein. Analog zum CYP-450-System existieren auch hier Familien und Subfamilien, deren Enzymaktivität durch Arzneistoffe moduliert wird.
mit Hilfe von Transportproteinen und Diffusionsprozessen können während des gesamten pharmakokinetischen Prozesses endogene und exogene Substanzen die biologischen Barrieren überwinden. der bekannteste Vertreter der wichtigsten Transporterfamilie, der „ATP-binding cassette transporter“ (ABC-Transporter), ist das P-Glykoprotein (P-gp). die physiologische Aufgabe von P-gp ist es, zu verhindern, dass toxische Substanzen in den systemischen Kreislauf oder über die Blut-Hirn-Schranke eindringen. Bereits resorbierte Stoffe können mittels dieses Transporters aus der Zelle wieder „herausgepumpt“ werden. P-gp wird deshalb auch als „Effluxpumpe“ bezeichnet. Ähnlich dem CYP-450ystem kann die Funktion von P-gp durch Arzneistoffe gehemmt oder induziert werden. Arzneistoffe, welche Substrate von cyp3a sind, sind oft auch Substrate von P-gp.
Literatur:
Pirmohamed M et al., Adverse drug reactions as cause of admission to hospital: prospective analysis of 18820 patients. BMJ 2004; 329:15–19.
Van Roon EN et al., Clinical relevance of drug-drug interactions. Drug Safety 2005; 28:1131–9.
Paine M, Kim RB, The human intestinal cytochrome P450 „pie“. Drug Metab Dispos 2006; 34:880–886.
Kiang TKL, Ensom MHH, Chang TKH, UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol & Therapeut 2005; 106:97–132.
Rosskopf D, Kroemer HK, Siegmund W, Pharmakokinetische Probleme in der Praxis. Rolle von Arzneimitteltransportern. Dt Med Wochenschr 2009; 134:345–56.
Thuerauf N, Fromm MF, The role of transporter P-glycoprotein for disposition and effects of centrally acting drugs and for the pathogenesis of cns diseases. Eur Arch Psychiatry Clin Neurosci 2006; 256:281–6.
Zafar A, Sharif MD, Pharmacokinetics, Metabolism and drug-drug interactions of atypical antipsychotics in special Populations. J Clin Psychiatry 2003; 5:22–25.
Spina E, De Leon J, Metabolic drug interactions with newer antipsychotics: a comparative review. Basic & Clinical Pharmacology & Toxicology 2007; 100:4–22.
Strobach D, Klinisch relevante Interaktionen zwischen Analgetika und Psychopharmaka. Arzneimitteltherapie 2012; 30:83–92.
Boyer EW, Shannon M, the serotonin syndrome. n engl J Med 2005; 352:1112–20.
Mittino D, Mula M, Monaco F, Serotonin syndrome associated with tramadol-sertralin coadministration. clin neuropharmacol 2004: 27:150–151.
Stephan P et al., Klonus, Hyperreflexie und Agitation bei einer Patientin mit hohem Fluvoxamin-serumspiegel: Symptome der Serotonin-Toxizität. Schweiz Med Forum 2008; 8:100–103.
Dunkley EJC et al. The Hunter SerotoninToxicity Criteria: simple and acurate diagnostic decision rules for serotonin toxicity. Q J Med 2003; 96:635–642.
Han L, Agostini JV, Allore HG, Cumulative anticholinergic exposure is associated with poor memory and executive function in older men. J AM Geriatr Soc 2008; 56:2203–10.
Rudolph JL et al., The anticholinergic risk scale and anticholinergic adverse effects in older persons. arch intern Med 2008; 168:508–13.
Delacrétaz E, Medikamente und verlängertes QT-Intervall. Schweiz Med Forum 2007; 7:814–819.
Haverkamp W, Haverkamp F, Breithardt G, Medikamentenbedingte Qt-Verlängerung und torsade de Pointes. deutsches Ärzteblatt 2002; 99:1972–1979.
De Abajo FJ, García-Rodríguez LA, risk of upper gastrointestinal tract bleeding associated with selective serotonin reuptake inhibitors and venlafaxine therapy: interaction with nonsteroidal antiinflammatory drugs and effect of acid-suppressing agents. Arch Gen Psychiatry 2008; 65:795–803.
Andrade C et al., serotonin reuptake inhibitor antidepressants and abnormal bleeding: a review for clinicians and a reconsideration of mechanisms. J Clin Psychiatry 2010; 71:1565–75.

AutorIn: Mag. Pharm. Sonja Mayer, AHPH
Klinische Pharmazeutin, Nervenklinik Wagner-Jauregg, Linz

Ursprünglich erschienen:
SP 04|2012
SP 04|2012