


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Rare and Undiagnosed Diseases: diagnostische Abklärung
22. Mai 2019
Der folgende Fall zeigt anschaulich, wie neue diagnostische Verfahren präzisere Diagnosen ermöglichen und dadurch zielgerichtete Therapieoptionen eingesetzt werden können:
Eine 34-jährige Patientin stellte sich mit einer palpablen Purpura mit Betonung der Extremitäten, teils urtikariellen Läsionen sowie schmerzhaften Erosionen der Mundschleimhaut vor (Abb. 1). Diese Symptome bestanden in wechselnder Ausprägung seit 8 Jahren. Außerdem berichtete die Patientin über tägliche Fieberschübe und Gelenkschmerzen. In der weiteren Abklärung fielen erhöhte Entzündungsparameter, eine Anämie und eine Splenomegalie auf. Aufgrund der Beteiligung der Mundschleimhaut war es innerhalb von 2 Jahren zu einem kompletten Zahnausfall gekommen. Bisherige Behandlungen – teils auch durch Hämatologen und Rheumatologen – inkludierten systemische und topische Glukokortikoide, Methotrexat, Azathioprin, Rituximab und verschiedene TNF-Blocker, die allesamt keine dauerhafte Verbesserung des Krankheitsbildes ergaben.In der Histopathologie konnte die klinische Verdachtsdiagnose einer Vaskulitis der kleinen Gefäße bestätigt werden, IgA-Ablagerungen waren in der Immunfluoreszenz nicht nachweisbar. Nachdem sämtliche mögliche Ursachen (Autoantikörper, Kollagenosen, bakterielle oder virale Infektionen, Kryoglobuline, Medikamente, Malignome) ausgeschlossen werden konnten, führten wir eine Abklärung mittels Genanalyse der über 1.000 häufigsten genetischen Ursachen für Immundefekte durch, die zwei heterozygote Mutationen des MEFV-Gens zeigte sowie eine zusätzliche, bisher unbekannte homozygote Mutation eines weiteren Gens.
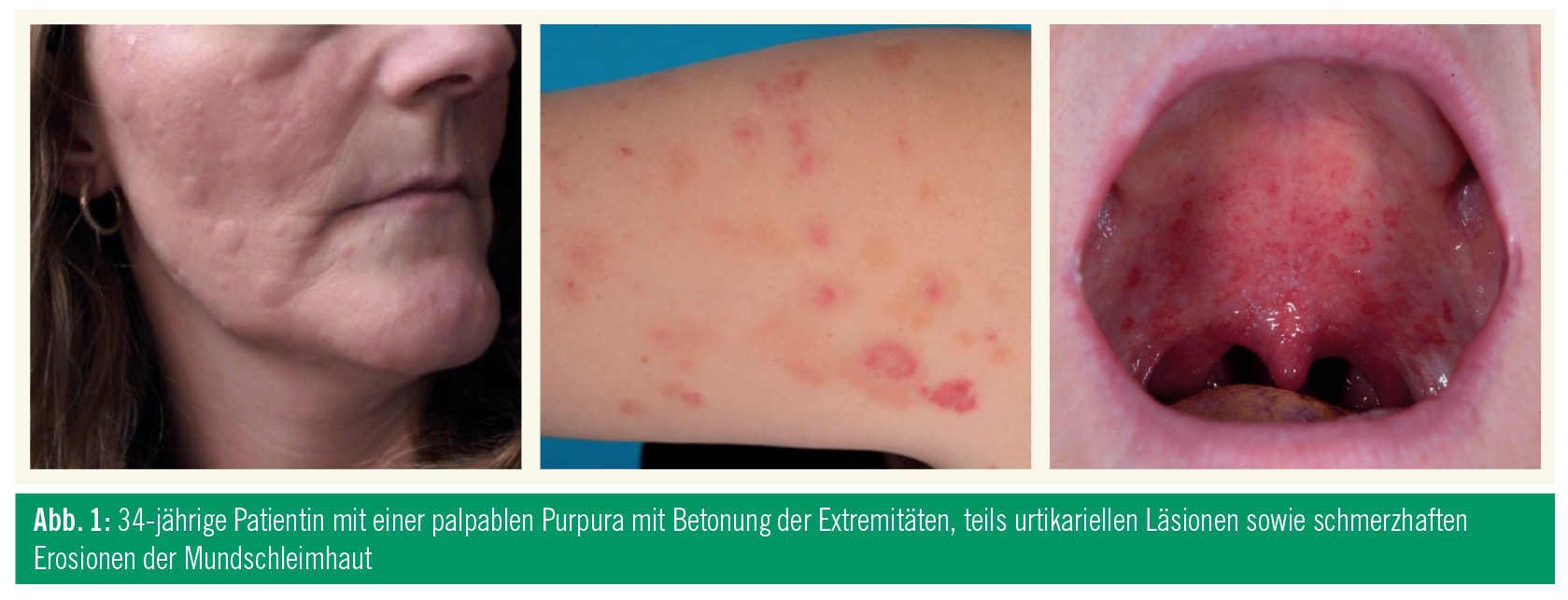
In der Folge stellten wir die Diagnose eines familiären Mittelmeerfiebers mit Hautbeteiligung im Sinne einer Vaskulitis. Zu erwähnen ist, dass auch heterozygote Mutationen zum familiären Mittelmeerfieber führen können, wenn weitere genetische Faktoren dazu beitragen. Dieser „second hit“ könnte durch die gefundene homozygote Mutation erklärbar sein. Die folgende Therapie mit dem Interleukin-1-Rezeptor-Antagonisten Anakinra führte zu einer Verbesserung der Entzündungsparameter, Fieberschübe, Gelenkschmerzen und der Anämie, allerdings mussten wir Anakinra aufgrund starker Lokalreaktionen absetzen und begannen stattdessen mit der Gabe von Colchizin. Während Entzündungsparameter, Fieberschübe und Anämie gut auf Colchizin ansprachen, bestanden die Affektionen der Haut und Schleimhäute weiter, sodass wir zusätzlich mit Dapson behandelten. Unter der Kombinationstherapie mit Dapson und Colchizin ist die Patientin nahezu erscheinungsfrei.
Seltene Erkrankungen – Definition und Bedeutung
Unter seltenen Erkrankungen – „rare diseases“ – fasst man definitionsgemäß jene Krankheiten zusammen, die nicht häufiger als bei einer von 2.000 Personen auftreten. Da etwa 8.000 seltene Erkrankungen beschrieben sind, könnten etwa 400.000 Menschen in Österreich von einer seltenen Erkrankung betroffen sein.
Durch neue diagnostische Möglichkeiten, die das Konzept der Präzisionsmedizin umschreiben, können seltene Erkrankungen besser und genauer diagnostiziert werden. Dies führt unweigerlich zu der Erkenntnis, dass selbst häufige Erkrankungen in Untergruppen aufgeteilt werden können und die Therapie auf den einzelnen Patienten maßgeschneidert werden sollte. Die Herausforderung an die moderne Diagnostik im Allgemeinen und speziell im Kontext der seltenen Erkrankungen liegt zweifellos in der Möglichkeit, breitenwirksam zu sein, aber dennoch auf die „Ausnahmen von der Regel“ Rücksicht zu nehmen.
Mit Breitenwirksamkeit ist die Durchführbarkeit im klinischen Alltag, also keine übermäßige Mehrbelastung des klinischen Personals, bei gleichzeitiger Kostenwirksamkeit für das Gesundheitswesen gemeint. Demnach müssen idente oder sehr ähnliche Analyseverfahren auf größere Patientengruppen angewendet werden. Ist diese erneute Standardisierung der natürliche Feind der Präzisionsmedizin? Die logische Konsequenz aus diesen Überlegungen ist eine wohlüberlegte Stratifizierung der Patienten auf Basis komplexerer und, durch moderne Techniken, präziserer Methoden und daraus resultierender Parameter. Das neue Schlüsselwort in diesem Kontext ist gar nicht mehr Präzisionsmedizin, sondern vielmehr die „High-Definition“-(HD-)Medizin. Darunter versteht man das genaue Beschreiben der Erkrankung auf molekularer Ebene im Zusammenspiel mit aufgezeichneten Lebensgewohnheiten des Patienten. Wie später noch erläutert wird, generieren bereits die Technologien der Gegenwart eine Menge an Daten, die über neuartige, computerbasierte Analysemethoden entschlüsselt werden müssen. Interdisziplinarität zwischen Klinik und Wissenschaft ist daher für die erfolgreiche Diagnosestellung der Zukunft entscheidend, insbesondere im Bereich der seltenen Erkrankungen.
Eine ganz andere, aber methodisch verwandte Herausforderung ist die Behandlung seltener Erkrankungen. Eine individuelle Therapie, die auf diagnostischen Methoden entsprechend dem jeweiligen Krankheitsbild beruht, ist das Ziel. Die größte Herausforderung im Ablauf all der Prozesse, die allein für die Diagnose notwendig wären, stellt die Dauer der Untersuchungen und deren Interpretation dar. Nur selten kann heute bei der Therapie auf diagnostische Parameter aus der HD-Medizin zurückgegriffen werden. Selbst bei erfolgreicher und präziser Diagnosestellung ist es oft sehr schwer, eine zielgerichtete Therapie anzuwenden.
Seltene Genodermatosen
Klinische Systematik: Während fast alle genetischen Erkrankungen auch selten sind, sind nicht alle seltenen Erkrankungen genetischen Ursprungs. Es gibt auch seltene infektiöse, autoimmunologische oder neoplastische Erkrankungen. Häufig ist die genaue Ursache einer seltenen Erkrankung unbekannt. Genodermatosen können aufgrund des molekularen Defekts oder basierend auf den betroffenen Körperregionen und klinischen Symptomen eingeteilt werden.
Alleine unter Genodermatosen gibt es etwa 500 verschiedene seltene Erkrankungen, die ein diverses klinisches Bild zeigen. So werden erbliche Verhornungsstörungen, die Epidermolysis-bullosa-hereditaria-Gruppe, vererbte Bindegewebsdefekte, erbliche neurokutane Syndrome sowie Erbkrankheiten mit erhöhtem Tumorrisiko und Hauterscheinungen bei primären Immundefizienzen unterteilt.
Diagnostik: Der gegenwärtige Goldstandard in der Diagnostik umfasst neben den altbewährten histologischen und immunhistochemischen Methoden auch die Polymerase-Kettenreaktion (PCR), welche im Grunde bereits als Vorstufe zu den neuen Technologien der letzten Generation im Bereich Genomsequenzierung gesehen werden kann. Während sich die PCR auf einzelne genomische Abschnitte beschränkt und deren Auswertung meist binär erfolgt (vorhandenes Produkt oder nichtvorhandenes Produkt), bietet die Genomsequenzierung ein umfassenderes Bild. Hierbei wird entweder das gesamte Genom sequenziert, oder es werden jene Bereiche des Genoms angereichert, die bereits im Kontext diverser Erkrankungen charakterisiert wurden. Letztere Methodik erhält rezent einen immer größeren Stellenwert, auch in der Diagnostik, da die Datenanalyse routinemäßig durchgeführt werden kann und keine größeren Ressourcen beansprucht und die Kosten für Sequenzierungen in den vergangenen Jahren stetig gefallen sind.
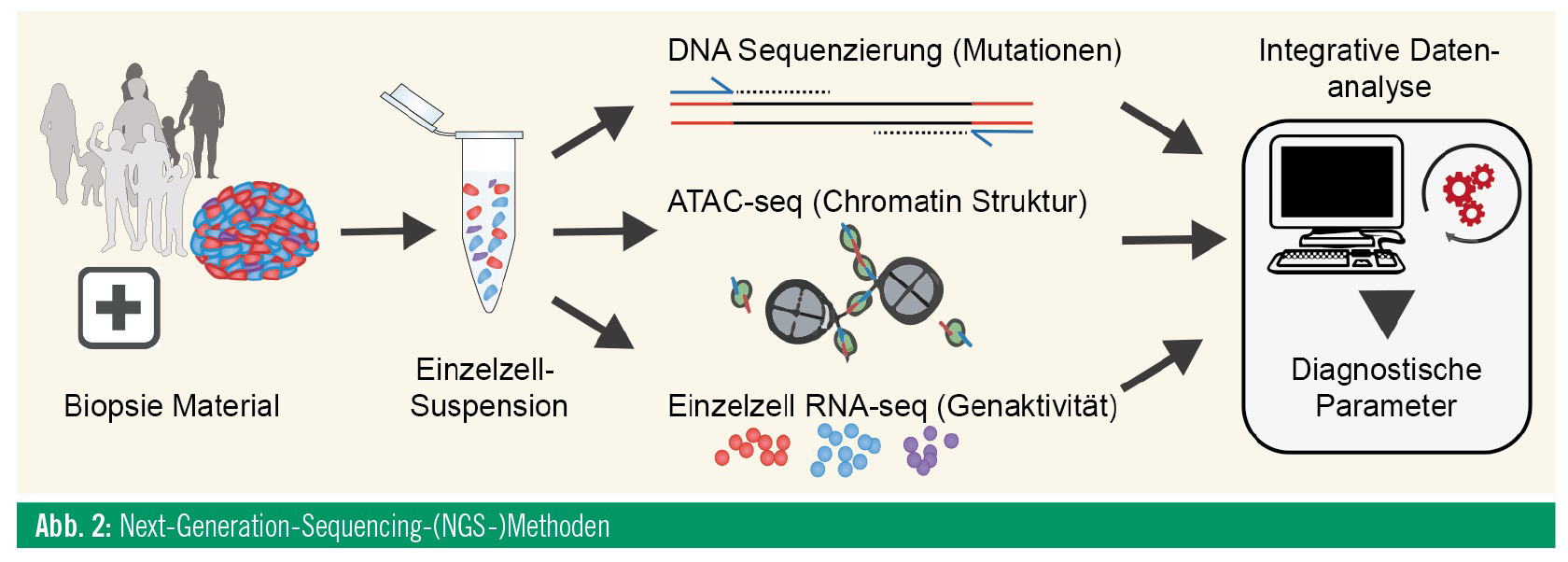
Neuere Entwicklungen im Bereich der Grundlagenforschung weisen in eine noch komplexere Zukunft. Die Entwicklung neuerer Methoden im Bereich der Sequenzierung und eine immer höhere Sensitivität der Verfahren, die mittlerweile bis auf die Ebene einzelner Zellen reicht, hat dazu geführt, dass dem Bereich der Epigenetik ein immer höherer Wert beigemessen wird. Wenn man sich die DNA als „Hardware“ vorstellt, dann wäre die Epigenetik die dazugehörige „Software“, die für die Regelung der Genaktivität in jeder einzelnen Zelle verantwortlich ist. Probleme der Hardware betreffen oftmals auch gleichzeitig die Software. Allerdings sind das Erkennen der Defekte und Zusammenhänge mit der vorliegenden Erkrankung im Falle der Epigenetik ungleich vielschichtiger. Die Techniken, die hier vermehrt zum Einsatz kommen, dienen zum Erkennen und Überprüfen der Schaltstellen entlang der DNA (Promotoren und Enhancer), welche für die Regulation der Genaktivität zuständig sind (Abb. 2). Wichtige Rollen spielen dabei Methylgruppen, die an diesen regulatorischen Sequenzen der DNA vorhanden sind, und die Chromatinstruktur als Maß für die Zugänglichkeit der regulatorischen Abschnitte der DNA. Erweitert um Einzelzellanalysen zur Darstellung der Heterogenität eines Tumors oder Gewebes, ermöglicht die kombinierte Anwendung dieser Techniken zum ersten Mal nicht nur eine Beschreibung der direkt aus Patienten gewonnenen molekularen Daten, sondern auch deren funktionelle Charakterisierung. Die Zusammenhänge der regulatorischen Prozesse können Ansatzpunkte für neue Medikamente sein, welche diese Prozesse je nach Bedarf beeinflussen können. Die Basis dieser Analysen und darauf aufbauender Interpretation der Daten bilden komplexe Computer-Algorithmen. Der Teilbereich der Bioinformatik, also jene Informatiker, die sich mit biologischen Daten und Themen befassen, bekommt daher in der Medizin und biomedizinischen Forschung der Zukunft einen immer höheren Stellenwert.
Aufgrund komplexer Datensätze begannen Wissenschafter und Mediziner in den letzten Jahren zu verstehen, dass erkrankte Zellen eines Tumors oder chronischer Entzündungsreaktionen in konstanter Wechselwirkung mit ihrer Umgebung stehen. Diese Umgebung wird vorwiegend von der Interaktion mit Immunzellen und anderen, strukturgebenden Zellen des Körpers geprägt. Die oben erwähnten Techniken zur Analyse der Heterogenität (beispielsweise von funktionell eingeschränkten Immunzellen innerhalb eines Tumors) und zur epigenetischen Charakterisierung könnten in Zukunft Antworten auf diverse seltene Erkrankungen der Haut liefern.
Bedeutung für die Dermatologie: Zwei Beispiele für solch einen Fall sind die Langerhans-Cell-Histiozytose (LCH) und das kutane T-Zell-Lymphom (CTCL). In beiden Fällen sind Immunzellen selbst – Makrophagen in der LCH und T-Zellen beim CTCL – der Ausgangszelltyp für die Krebserkrankung. Die LCH tritt zumeist im frühen Kindesalter auf (noch vor dem 2. Lebensjahr), wohingegen das CTCL meistens bei älteren Erwachsenen diagnostiziert wird. Beide Erkrankungen sind durch eine hohe He
terogenität sowohl auf Patientenebene als auch innerhalb der Erkrankungen selbst geprägt. So gibt es neben der Haut auch ein Auftreten der LCH u. a. am Knochen und beim CTCL im peripheren Blut (Sézary-Syndrom). In beiden Fällen kann man jedoch von einer starken Beteiligung des umgebenden Gewebes und der infiltrierenden gesunden Immunzellen ausgehen, die über entzündliche Prozesse auf die Epigenetik der Krebszellen einwirken und somit auf deren Entwicklung Einfluss nehmen. Während es für die LCH derzeit in über 80 % der Fälle gute Aussichten auf eine erfolgreiche Therapie gibt, ist eine kurative Therapie für das CTCL noch nicht in greifbarer Nähe. Eine genaue beschreibende und funktionelle Aufarbeitung der Erkrankungen unter Zuhilfenahme der zuvor erwähnten Sequenziermethoden könnte in Zukunft neue Ansatzpunkte für die Diagnostik, Patientenstratifizierung und auch Therapie liefern. Der Zugang zu geeignetem Patientenmaterial und Techniken für die entsprechende Aufarbeitung ist hier von immanenter Bedeutung. Genaue Planung und Organisation der anzuwendenden Techniken in Abhängigkeit vom meist limitierten Patientenmaterial ist hier der Schlüssel zum Erfolg.
Skin-RUD-Programm: Um den beschriebenen Herausforderungen bestmöglich zu begegnen, wurde an der Universitätsklinik für Dermatologie der Medizinischen Universität Wien eine interdisziplinäre Task Force gegründet. Das neue Skin-RUD-(Rare and Undiagnosed Diseases-)Programm unter der Leitung von Assoz. Prof. Dr. Georg Stary nimmt Zuweisungen von Patienten mit unklaren Hauterscheinungen gerne entgegen (Terminvereinbarung unter georg.stary(at)meduniwien.ac.at) und wird diese nach entsprechender Abklärung gemeinsam mit den betreuenden Ärzten besprechen.

AutorIn: Mag. Dr. Matthias Farlik, PhD
Universitätsklinik für Dermatologie, Medizinische Universität Wien;Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases (LBI-RUD);CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences

AutorIn: Assoc. Prof. Priv.-Doz. Dr. Georg Stary
Universitätsklinik für Dermatologie, Medizinische Universität Wien;Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases (LBI-RUD);CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences
Foto: MedUni Wien/Matern

Ursprünglich erschienen:
SD 02|2019
SD 02|2019
Herausgeber: Ao. Univ.-Prof. Dr. Christoph Höller, Assoc. Prof. Priv.-Doz. Dr. Constanze Jonak, Univ.-Prof. Dr. Rainer Kunstfeld, Univ.-Prof. Dr. Hubert Pehamberger
Publikationsdatum: 2019-05-22
Zur Ausgabe »
Publikationsdatum: 2019-05-22
Zur Ausgabe »
