


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hemmkörper-Entwicklung bei Patienten mit leichter Hämophilie A
16. Juli 2012
Hämophilie A ist eine angeborene, X-chromosomal rezessiv vererbte Gerinnungsstörung, welcher ein Mangel bzw. das Fehlen des Gerinnungsfaktors VIII (FVIII) zugrunde liegt. Die Inzidenz liegt bei etwa 1:5.000 in der männlichen Bevölkerung. Abhängig von der FVIII-Aktivität werden unterschiedliche Schweregrade unterschieden: Patienten mit einer FVIII-Aktivität (FVIII:C) unter 1 % haben eine schwere Hämophilie A, Patienten mit FVIII:C zwischen 1 und 5 % eine mittelschwere Form und jene mit FVIII:C zwischen 5 und 40 % eine leichte Form der Hämophilie.1
Abhängig vom Schweregrad zeigen die Patienten eine unterschiedliche Blutungsneigung: während schwere Hämophilie zu spontanen Blutungen führt und daher meist in der frühen Kindheit diagnostiziert wird, haben Patienten mit leichter Hämophilie kaum Spontanblutungen. Daher wird bei diesen die Diagnose auch oft erst im Erwachsenenalter gestellt.
Eine die Therapie von Hämophiliepatienten erschwerende Komplikation ist das Auftreten eines Hemmkörpers gegen den FVIII. Solche Inhibitoren treten bei schwerer Hämophilie sehr häufig (bei 20–30 % der Patienten) auf.2, 3
Bei leichter Hämophilie liegt die Hemmkörper- Inzidenz etwa bei 3–13 %.4, 5 Das Auftreten eines Inhibitors ändert allerdings den Blutungsphänotyp in einen schweren und stellt somit eine große Herausforderung für den Behandler dar.
Üblicherweise treten Hemmkörper bei leichten Hämophiliepatienten nach intensiver Behandlung auf, sind niedrigtitrig und transient.
An der Gerinnungsambulanz des AKH Wien werden 213 Erwachsene mit Hämophilie behandelt, 58 % davon mit leichter Hämophilie.
In den letzten 30 Jahren trat bei nur zwei Patienten mit leichter Hämophilie ein hoch – titriger Hemmkörper auf.

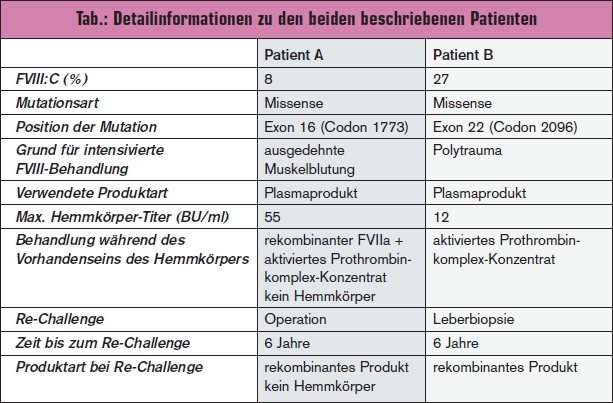
Patient A
Die Diagnose einer leichten Hämophilie A wurde im Volksschulalter gestellt. Als zugrunde liegende Mutation wurde eine Missense-Mutation im Exon 16, Codon 5375 (T>C-Transition, Val1773Ala) detektiert. Diese Mutation ist in der Hämophiliemutationsdatenbank HABD6 bis dato nicht be – schrieben. Der Patient erhielt im Alter von 34 Jahren erstmals ein Faktorkonzentrat (FVIII angereicherte Plasmafraktion niederer Reinheit) nach einer traumatischen Ellenbogenblutung. Im Alter von 39 Jahren bekam er erneut ein Faktorkonzentrat im Rahmen einer Hüftgelenksoperation und im Alter von 41 Jahren aufgrund einer Magenblutung. Mit 47 Jahren erhielt der Patient eine Hüftgelenksprothese links und wurde in diesem Zusammenhang mit einem Faktorkonzentrat intermediärer Reinheit behandelt. Auch diese Operation verlief komplikationslos.
Im Alter von 50 Jahren hatte der Patient eine ausgedehnte spontane Muskelblutung, welche mit großen Mengen eines plasmatischen FVIII-Produktes behandelt wurde (Immunate®). Nach zwei Tagen intensiver Behandlung war die Wirkung nicht adäquat, der messbare FVIII-Spiegel lag unter 1 % und es wurde ein Hemmkörper von 3,5 Bethesda-Einheiten (BU)/ml gemessen, welcher im Verlauf einen max. Titer von 55 BU/ml erreichte. Die Muskelblutung wurde erfolgreich mit aktiviertem Prothrombinkomplex-Konzentrat (Feiba®) und aktiviertem Faktor VII (Novoseven®) behandelt. Der Inhibitor- Titer sank ohne Eradikationstherapie und war schließlich nicht mehr nachweisbar. Drei Jahre nach Auftreten des Hemmkörpers erhielt der Patient neuerlich große Mengen FVIII („Re-Challenge“) aufgrund einer Hüftoperation sowie 2 Jahre später im Rahmen einer urologischen Operation. Beide Male wurde Kogenate® verabreicht; es kam zu keinem Wiederauftreten des Hemmkörpers. Auch bei einer weiteren Exposition ein Jahr später (Synovektomie und Hüftoperation) wurde Kogenate® gegeben. Die FVIII-Antwort war adäquat und es war kein Inhibitor messbar (Details > Tab.).
Patient B
Bei Patient B wurde die Diagnose leichte Hämophilie A auf Basis einer bis dahin nicht beschriebenen Missense-Mutation im Exon 22 (c.6344T>C, Leu2096Pro) im Alter von 22 Jahren gestellt, als es nach Zahnextraktion zu einer deutlich verlängerten Nachblutung kam. Mit 28 Jahren erfolgte die erste Behandlung mit einem Faktorkonzentrat von intermediärer Reinheit. Im Alter von 50 Jahren hatte der Patient aufgrund eines retroperitonealen Hämatoms eine Operation und erhielt ein plasmatisches FVIII-Produkt. Ein Jahr danach hatte er ein Polytrauma mit starken Blutungen und erhielt Haemate®. Nach 3 Tagen Behandlung war der Faktoranstieg nicht adäquat, der FVIII-Spiegel lag unter 1 % und es wurde ein Hemmkörper mit einem maximalen Titer von 12 BU/ml gemessen. Der Patient wurde in der Folge mit Feiba® therapiert und erhielt keine Eradikationstherapie. Der Hemmkörper verschwand spontan und war nach 2 Monaten nicht mehr messbar. 6 Jahre später wurde eine Leberbiopsie durchgeführt, bei welcher der Patient ein rekombinantes FVIII-Produkt erhielt. Der FVIII-Anstieg war adäquat und der Hemmkörper trat nicht wieder auf (Details > Tab.).
RESÜMEE: Bei beiden beschriebenen Fällen hat sich nach intensiver FVIII-Gabe ein hochtitriger Hemmkörper entwickelt. Dieser verschwand jeweils spontan und bei einem Re- Challenge, d. h. erneuter FVIII-Gabe einige Jahre danach, kam es zu keinem Wiederauftreten des Inhibitors. Es wurde bereits von Hay et al.7 beschrieben, dass bei leichten Formen der Hämophilie hochtitrige Hemmkörper spontan verschwinden können, was durch eine Art Toleranzeffekt durch den endogenen FVIII, welcher ja kontinuierlich vorhanden ist, erklärt wurde. Dies und die Tatsache, dass Immuntoleranztherapie bei Hämophilie-Patienten mit Hemmkörperentwicklung sowohl kostspielig als auch anstrengend für den Patienten und oftmals nicht erfolgreich ist, führt uns zu dem Schluss, dass eine „Watch and wait“-Strategie durchaus angewandt werden kann. Blutungen können zwischenzeitlich (bis zum Verschwinden des Hemmkörpers) erfolgreich mit Prothrombinkomplex-Konzentrat bzw. aktiviertem Faktor VII behandelt werden.
FACT-BOX
• Hemmkörper-Entwicklung bei Patienten mit leichter Hämophilie A, insbesondere
das Auftreten hochtitriger Inhibitoren, ist eine seltene Komplikation, v. a. nach
intensiver FVIII-Substitution.
• Die Hemmkörper verschwinden oftmals spontan, Blutungen können zwischenzeitlich gut mit „bypassing agents“ (Feiba®, Novoseven®) therapiert werden,
sodass eine Immuntoleranztherapie kritisch hinterfragt werden sollte.
• In vielen Fällen ist eine „Watch and wait“-Strategie sicher eine überlegenswerte Alternative.

Ursprünglich erschienen:
UIM 02|2012
UIM 02|2012
Herausgeber: Univ.-Prof. Dr. Günter J. Kreijs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2012-03-09
Zur Ausgabe »
Publikationsdatum: 2012-03-09
Zur Ausgabe »
