


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
JAK-Inhibitoren („-citinibe“) – Entwicklung und Wirkmechanismus
28. November 2019
Die Therapie der rheumatoiden Arthritis (RA) hat sich in den vergangenen Jahrzehnten, vor allem durch die Einführung der Biologika, wesentlich verbessert. Davor wurde das primäre Therapieziel der RA, die Remission (≥ 6 Monate), durch konventionelle krankheitsmodifizierende Antirheumatika (engl. Abkürzung: cDMARDs) nur bei ca. 20 % der Patienten erreicht.1 Mit Unterstützung der Biologika erreichen mittlerweile bis zu 75 % der RA-Patienten die Remission.2
Durch die Entwicklung und Zulassung der Januskinase-Inhibitoren („-citinibe“) im Laufe der 2010er-Jahre wurden drei weitere Fortschritte in der RA-Therapie erreicht:
- Durch den neuen Wirkmechanismus erreichen noch mehr RA-Patienten die Remission.
- JAK-Inhibitoren werden nicht wie die Biologika parenteral, sondern oral verabreicht und haben im Körper nur eine kurze Verweildauer.
- Im Gegensatz zu den Biologika tritt bei JAK-Inhibitoren nur selten ein sekundärer Wirkverlust auf.
Der JAK/STAT-Signalweg
Der JAK/STAT-Signalweg (Abb.) ist eine wichtige Komponente vieler Zytokin- und Hormonrezeptor-Systeme, die Wachstum, Überleben, Differenzierung und Proliferation von Säugetierzellen regulieren. Darüber hinaus spielt er eine Schlüsselfunktion bei hämatologischen, immunologischen und chronisch entzündlichen Erkrankungen, die durch Zytokine vorangetrieben werden.

Januskinasen, kurz JAKs, sind für Zytokin-Rezeptoren wichtige Adaptermoleküle, um intrazelluläre Signalkaskaden zu aktivieren. Januskinasen wurden nach dem doppelköpfigen römischen Gott Janus, dem Wächter der Tore, benannt, da sie zwei ähnliche Proteindomänen haben, von denen aber nur eine funktional ist. Zur JAK-Familie zählen die vier Tyrosinkinasen JAK1, JAK2, JAK3 und TYK2. Diese liegen als Dimere vor, die sich entweder aus zwei gleichen oder aus zwei verschiedenen Bestandteilen zusammensetzen. Das Andocken eines Zytokins an seinen Rezeptor induziert die Rezeptordimerisierung, wodurch die assoziierten JAKs aktiviert werden und sich selbst und den Rezeptor an Tyrosinresten phosphorylieren. Die übertragenen Phosphatgruppen stammen dabei von ATP-Molekülen. Diese phosphorylierten Stellen dienen als Andockstellen für die Src-Homology-2-(SH2-)Domänen der „Signaltransduktoren und Aktivatoren der Transkription“ (STATs). Darauf folgt die Phosphorylierung und Bindung zweier STATs, welche als Dimere in den Zellkern wandern und als Transkriptionsfaktoren die Expression eines Zielgenes induzieren, wodurch Entzündungsreaktionen hervorgerufen oder verstärkt werden können.
Wirkmechanismus der JAK-Inhibitoren
JAK-Inhibitoren, die chemisch den kleinen Pyrrolopyrimidinen entsprechen, ähneln in ihrer Struktur dem ATP und binden reversibel an die ATP-Bindungsstelle der JAKs. Dadurch verhindern sie die Übertragung einer Phosphatgruppe auf die JAK-Moleküle und somit die Signaltransduktion (Abb.). JAK-Inhibitoren fangen im Gegensatz zu den Biologika die Zytokinsignale nicht im Extrazellulärraum, sondern intrazellulär ab und hemmen daher nicht nur ein bestimmtes Zytokin, sondern gleich mehrere. Da die komplette Blockade multipler Zytokine zu einer erheblichen Infektneigung führen würde, werden die JAK-Inhibitoren so dosiert, dass die Wirkung der meisten Zytokine nur um ca. die Hälfte reduziert wird. Die Zytokin-Rezeptoren nutzen nicht alle vier JAKs für die Signaltransduktion, sondern in der Regel nur zwei. Die derzeit in der Therapie der RA zugelassenen JAK-Inhibitoren Tofacitinib (Xeljanz®) und Baricitinib (Olumiant®) haben unterschiedliche Affinitäten zu den verschiedenen JAKs: Tofacitinib inhibiert hauptsächlich JAK1 und JAK3, Baricitinib hemmt v. a. JAK1 und JAK2. Durch diese unterschiedlichen Präferenzen sind die durch Tofacitinib und Baricitinib beeinflussten Zytokine nicht komplett ident.
Entwicklung und Zulassung im Zeitraffer (Tab.)
Die Vorgeschichte der JAK-Inhibitoren begann mit der Erstbeschreibung der Tyrosinkinasen JAK1, JAK2 und TYK2 in den Jahren 1989–1991 (zusammengefasst von Stark GR und Darnell JE Jr.)3, gefolgt von der Entdeckung der vierten Kinase, JAK3, im Jahr 1994. Wesentliche Hinweise auf die Bedeutung des JAK-Signalwegs für Zellentwicklung, Überleben und Differenzierung brachten ab 1995 die ersten In-vivo-Studien an JAK-Knock-out-Mäusen, die entweder einen stark beeinträchtigten Phänotyp aufwiesen oder nicht lebensfähig waren. In ähnlicher Weise führen JAK3-Mutationen beim Menschen zum autosomal rezessiven Immundefizienzsyndrom („X-linked severe combined immunodeficiency“, SCID). Januskinasen sind somit für die Funktionalität der Signaltransduktionswege innerhalb der Zelle essenziell. Die Überexpression einzelner Komponenten des JAK-STAT-Signalwegs führt hingegen zu überschießenden Immun- und Autoimmunreaktionen. 2006 wurde eine erhöhte Expression von STAT1, STAT4 und JAK3 bei Patienten mit entzündlicher Arthritis festgestellt.4 Weitere Beispiele für Krankheiten, bei denen der JAK-Signalweg überaktiv ist, sind chronisch entzündliche Darmerkrankungen, Atherosklerose, neurodegenerative Krankheiten, einige solide Tumoren sowie Asthma und Allergien.
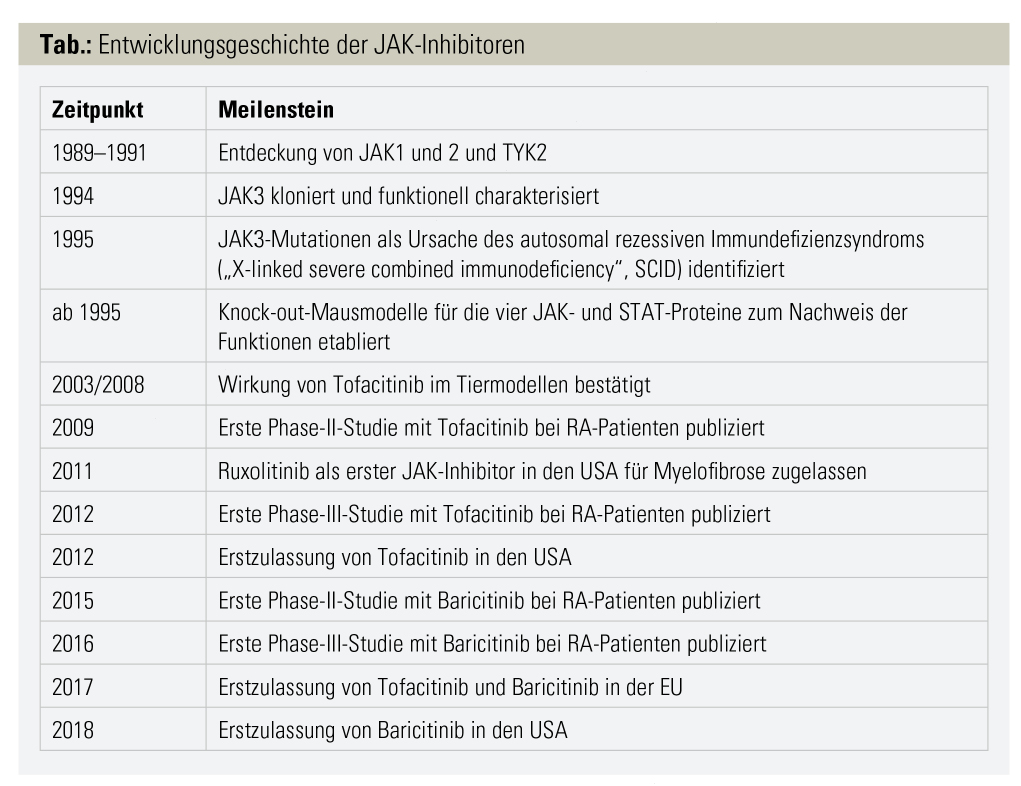
2003 wurde erstmals die immunsupprimierende Wirkung des JAK-Inhibitors Tofacitinib (CP-690550) in einem vorklinischen, tierischen Transplantationsmodell beschrieben.5 Fünf Jahre später publizierten Milici et al. die positiven Effekte von Tofacitinib in einem Arthritis-Tiermodell.6 Die Wirksamkeit von Tofacitinib bei RA-Patienten, die zuvor unzureichend auf cDMARDs und Biologika angesprochen hatten, wurde 2009 in einer Phase-IIa-Studie von Kremer et al. bestätigt.7 Entsprechende Phase-III-Studien, wie die ORAL-Solo-Studie von Fleischmann et al.8 oder die ORAL-Standard-Studie von van Vollenhoven et al.9, untermauerten den Erfolg von Tofacitinib bei RA-Patienten und wurden ab 2012 veröffentlicht (ORAL Scan: 201310, ORAL Step: 201311, ORAL Start: 201412, ORAL Sync: 201813, ORAL Strategy: 201914). Die erste Phase-II-Studie zur Wirksamkeit von Baricitinib bei RA-Patienten, die zuvor unzureichend auf Methotrexat (MTX) angesprochen hatten, folgte erst 2015.15 Ein Jahr später erschien mit der RA-BEACON-Studie16 auch die erste Phase-III-Studie zur Wirksamkeit von Baricitinib bei therapierefraktären RA-Patienten17 (RA-BEGIN: 201718, RA-BEAM: 201719, RA-BUILD: 201720).
Im Jahr 2011 wurde in den USA Ruxolitinib als erster JAK-Inhibitor für die Therapie von Patienten mit Myelofibrose zugelassen. Im November 2012 folgte die FDA-Zulassung des JAK-Inhibitors Tofacitinib für die Therapie erwachsener Patienten mit moderater bis schwerer aktiver RA, die nicht ausreichend auf MTX angesprochen oder dieses nicht vertragen hatten. Seit 2013 ist Tofacitinib auch für RA-Patienten in der Schweiz zugänglich. Für die EU wurde die Zulassung bereits 2011 beantragt, aber im April 2013 zunächst abgelehnt. Erst 2017 wurden Tofacitinib und Baricitinib für die Therapie der mittelschweren bis schweren RA bei erwachsenen Patienten, die auf ein oder mehrere cDMARDs unzureichend angesprochen oder diese nicht vertragen hatten, auch in der EU zugelassen.
Sowohl in den europäischen als auch in den deutschen Leitlinien zur Therapie der RA werden JAK-Inhibitoren als gleichrangige Alternative zu den Biologika empfohlen. Tofacitinib und Baricitinib sind in der Kombination mit MTX und in der Monotherapie zugelassen.
Klinisches Ansprechen
Gegen Biologika können auch nach Jahren der erfolgreichen Therapie neutralisierende Anti-Drug-Antikörper gebildet werden, die zu einem Wirkverlust führen. Im Gegensatz dazu werden gegen die kleinen JAK-Inhibitoren keine Anti-Drug-Antikörper gebildet – der einmal erreichte Therapieerfolg hält somit an. Studienergebnisse attestieren den JAK-Inhibitoren nicht nur eine langanhaltende Wirksamkeit, sondern im Vergleich zu den Biologika auch einen schnellen Wirkeintritt.
Tofacitinib: In allen ORAL-Phase-III-Studien zeigten die mit zweimal täglich Tofacitinib behandelten RA-Patienten im Vergleich zu den mit Placebo (oder cDMARDs oder MTX) behandelten Patienten statistisch signifikant höhere Ansprechraten. In der ORAL-Solo-Studie8 wurden bei einer Gabe von 2-mal täglich 10 mg Tofacitinib nach 6 Monaten folgende Ansprechraten vermerkt: ACR20*: 71 %, ACR50: 47 % und ACR70: 29 %. In der ORAL-Start-Studie12 hatten Patienten, die 2-mal täglich mit 10 mg Tofacitinib behandelten wurden, nach 6 Monaten deutlich höhere Ansprechraten (ACR20: 75 %, ACR50: 56 %, ACR70: 37 %) als Patienten, die mit MTX behandelt wurden (ACR20: 51 %, ACR50: 27 %, ACR70: 12 %). In der ORAL-Sync-Studie13 lagen die Ansprechraten auf Tofacitinib + cDMARDs bei ACR20: 57 %, ACR50: 33 % und ACR70: 16 %, hingegen auf Placebo + cDMARDs nur bei ACR20: 31 %, ACR50: 13 % und ACR70: 3 %. Während des Verlaufs der Studie ORAL Strategy14 sprachen in der Gruppe mit 2-mal täglich 5 mg Tofacitinib + MTX numerisch ähnlich viele Patienten auf die Behandlung an wie in der Gruppe mit 40 mg Adalimumab + MTX (Tofacitinib + MTX: ACR20: 73 %, ACR50: 46 %, ACR70: 25 % versus Adalimumab + MTX: ACR20: 70 %, ACR50: 43 %, ACR70: 20 %). In der ORAL-Standard-Studie9 zeigten Patienten, die 2-mal täglich mit 10 mg Tofacitinib + MTX behandelt wurden, sogar bessere Ansprechraten als Patienten der Adalimumab+ MTX-Gruppe (Tofacitinib + MTX: ACR20: 51 %, ACR50: 34 %, ACR70: 21 % versus Adalimumab + MTX: AC20: 46 %, ACR50: 27 %, ACR70: 9 %). In der ORAL-Scan-Studie10 mit Patienten, die inadäquat auf MTX angesprochen hatten (ACR20: 25 %, ACR50: 8 %, ACR70: 1 %), wurden bei einer Gabe von 2-mal täglich 10 mg Tofacitinib + MTX nach 6 Monaten folgende Ansprechraten vermerkt: ACR20: 62 %, ACR50: 44 % und ACR70: 22 %. Für die ORAL-Step-Studie11 mit Patienten, die mit Tumornekrosefaktor-(TNF-)α-Blockern nicht ausreichend versorgt waren, wurden für Patienten, die für 3 Monate 2-mal täglich 10 mg Tofacitinib + MTX erhielten (ACR20: 48 %, ACR50: 28 %, ACR70: 10 %) bessere Ansprechraten ermittelt als für Patienten, die nur noch MTX erhielten (ACR20: 24 %, ACR50: 8 %, ACR70: 2 %).
Baricitinib: In allen Phase-III-Studien mit Baricitinib hatten jene Patienten, die 4 mg Baricitinib 1-mal täglich erhielten, nach 12 Wochen statistisch signifikant höhere ACR20-, ACR50- und ACR70-Ansprechraten als Patienten mit Placebo, MTX oder Adalimumab. Die Wirksamkeit trat für alle Parameter rasch ein, wobei ein signifikant stärkeres Ansprechen bereits nach Woche 1 beobachtet wurde. Ein statistisch signifikant größerer Anteil der mit Baricitinib 4 mg behandelten Patienten erreichte im Vergleich zu jenen mit Placebo oder MTX eine Remission, definiert als SDAI** ≤ 3,3 und CDAI*** ≤ 2,8, in den Wochen 12 und 24. In der RA-BEGIN-Studie18 lag die ACR20-Ansprechrate für 1-mal täglich 4 mg Baricitinib nach 12 Wochen bei 79 %, die von MTX bei nur 59 % und die der Kombination aus Baricitinib + MTX bei 77 %. Folgende ACR50-Ansprechraten wurden erfasst: Baricitinib: 55 %, MTX: 33 % und Baricitinib + MTX: 60 %. Bei Patienten, die Baricitinib erhielten, war die ACR70-Response bei 31 %, bei Patienten, die nur mit MTX behandelt wurden, ergab sich eine ACR70-Response von 16 %, bei Patienten, die eine Kombination aus Baricitinib und MTX erhielten, wurde eine ACR70-Response von 34 % beobachtet. In der RA-BEAM-Studie19 mit Patienten, die zuvor inadäquat auf MTX angesprochen hatten, wurden nach 12 Wochen Therapie mit einmal täglich 4 mg Baricitinib folgende Ansprechraten ermittelt: ACR20: 70 %, ACR50: 45 % und ACR70: 19 %. Die Ansprechraten für die Vergleichsgruppe mit Adalimumab waren deutlich niedriger (ACR20: 61 %, ACR50: 35 %, ACR70: 13 %). Auch in den Studien RA-BUILD20 (ACR20: 62 %, ACR50: 34 %, ACR70: 18 %) und RA-BEACON16 (ACR20: 55 %, ACR50: 28 %, ACR70: 11 %) mit Patienten, die inadäquat auf cDMARDs bzw. TNF-α-Blocker angesprochen hatten, wurden für 1-mal täglich 4 mg Baricitinib nach 12 Wochen deutlich bessere Ansprechraten erzielt als in den Placebogruppen.
Aufgrund der relativ kurzen Halbwertszeit von einigen Stunden (Tofacitinib: 3 h, Baricitinib: 13 h) ist die Therapie mit JAK-Inhibitoren auch gut steuerbar, und der Wirkstoff kann bei Infekten, geplanten operativen Eingriffen oder einem Kinderwunsch rasch ausgewaschen werden.
Ausblick
Die von JAK-Inhibitoren beeinflussten Zytokine spielen auch in der Pathogenese anderer Erkrankungen, wie chronisch entzündlichen Darmerkrankungen, Kollagenosen, Vaskulitiden und Asthma, eine wichtige Rolle. Es ist daher in Zukunft mit weiteren Studien und Zulassungserweiterungen für die JAK-Inhibitoren zu rechnen. Zudem werden neue JAK-Inhibitoren entwickelt und geprüft, die spezifischer einzelne JAKs blockieren, ein geringeres Nebenwirkungsspektrum aufweisen und selektiver einzelne Gruppen von Zytokinen inhibieren können. Die Ergebnisse der ersten Phase-III-Studien dieser „New generation“-JAK-Inhibitoren bei RA-Patienten wurden bereits im Laufe der letzten zwei Jahre präsentiert.
In den 2018 veröffentlichten Phase-III-Studien SELECT-BEYOND21 und SELECT-NEXT22 wurde der JAK1-Inhibitor Upadacitinib bei RA-Patienten mit inadäquatem Ansprechen auf DMARDs und Biologika erfolgreich als Add-on-Medikation geprüft. Auch in der 2019 publizierten SELECT-MONOTHERAPY-Studie war Upadacitinib als Monotherapie bei RA-Patienten mit inadäquatem Ansprechen auf MTX der Placebotherapie signifikant überlegen.23 Im Juli 2019 wurde in der FINCH-2-Studie gezeigt, dass der JAK1-Inhibitor Filgotinib bei RA-Patienten unter DMARD-Therapie mit inadäquatem Ansprechen auf Biologika besser wirksam war als die Placebotherapie.24 Auswertungen weiterer randomisierter Studien mit Filgotinib sind bereits im Gange. Ebenfalls im Juli 2019 wurden die Daten der Studie RAJ4 veröffentlicht. In dieser Studie wurde der JAK1-Inhibitor Peficitinib bei RA-Patienten mit inadäquatem Ansprechen auf MTX geprüft und zeigte ebenso eine Überlegenheit gegenüber Placebo.25
Resümee
JAK-Inhibitoren und Biologika bieten auf Basis der bisherigen Studiendaten eine vergleichbare Wirksamkeit und Sicherheit in der Behandlung der RA. In der Praxis wird der JAK-Inhibitor in Form einer ein- bzw. zweimal täglichen Tablette oft einer Biologika-Injektion oder -Infusion vorgezogen. Allerdings sind die Langzeitdaten zu den JAK-Inhibitoren bezüglich Tumorrisiko, kardiovaskulären Ereignissen und Mortalität noch nicht ausreichend. Mindestens 10–15 Jahre Therapieerfahrung werden nötig sein, um ein erhöhtes Risiko vollständig ausschließen zu können.
* Die ACR-Response-Kriterien bewerten, ob es durch die Therapie zu einer Besserung von vordefinierten Symptomen wie Gelenkschmerz, Gelenkschwellung oder Funktionsbeeinträchtigung gekommen ist. Eine mindestens 20%-Verbesserung entspricht einer ACR20-Response (ACR20).
** Ein Instrument zur Beurteilung der Krankheitsaktivität ist der sogenannte „Simplified Disease Activity Index”, abgekürzt SDAI. Der SDAI beruht auf der Untersuchung von 28 Gelenken und der CRP-Bestimmung. Beim SDAI entsprechen Werte unter 3,3 einer Remission.
*** Lässt man den Laborwert CRP weg, kommt man zu dem einfach zu errechnenden „Clinical Disease Activity Index“, abgekürzt CDAI. Beim CDAI entsprechen Werte unter 2,8 einer Remission.
1 Eberhardt K et al., Br J Rheumatol 1998; 37(12):1324–9
2 Lauter A et al., Z Rheumatol 2019; 78(7):660–9
3 Stark GR et al., Immunity 2012; 36(4):503–14
4 Walker JG et al., Ann Rheum Dis 2006; 65(2):149–56
5 Changelian PS et al., Science 2003; 302(5646):875–8
6 Milici AJ et al., Arthritis Res Ther 2008; 10(1):R14
7 Kremer JM et al., Arthritis Rheum 2009; 60(7):1895–905
8 Fleischmann R et al., N Engl J Med 2012; 367(6):495–507
9 van Vollenhoven RF et al., N Engl J Med 2012; 367(6):508–19
10 van der Heijde D et al., Arthritis Rheum 2013; 65(3):559–70
11 Burmester GR et al., Lancet 2013; 381(9865):451–60
12 Lee EB et al., N Engl J Med 2014; 370(25):2377–86
13 Li Z-G et al., Chin Med J (Engl) 2018; 131(22):2683–92
14 Calabrese LH et al., Arthritis Care Res (Hoboken) 2019; DOI: 10.1002/acr.24010 [Epub ahead of print]
15 Keystone EC et al., Ann Rheum Dis 2015; 74(2):333–40
16 Genovese MC et al., Rheumatology (Oxford) 2018; 57(5):900–8
17 Genovese MC et al., N Engl J Med 2016; 374(13):1243–52
18 van der Heijde D et al., Clin Rheumatol 2018; 37(9):2381–90
19 Taylor PC et al., J Clin Med 2019;8(6 ). pii: E831. DOI: 10.3390/jcm8060831
20 Dougados M et al., Ann Rheuma Dis 2017; 76(1):88–95
21 Genovese MC et al., Lancet 2018; 391(10139):2513–24
22 Burmester GR et al., Lancet 2018; 391(10139):2503–12
23 Smolen JS et al., Lancet 2019; 393(10188):2303–11
24 Genovese MC et al., JAMA 2019; 322(4):315–25
25 Takeuchi T et al., Ann Rheum Dis 2019; 78(10):1305–19

AutorIn: Assoz. Prof. Priv.-Doz. Dr. Eva Sturm
Otto Loewi Forschungszentrum für Gefäßbiologie, Immunologie und Entzündung, Lehrstuhl für Pharmakologie, Medizinische Universität Graz

AutorIn: Univ.-Prof. Dr. Akos Heinemann
Otto Loewi Forschungszentrum für Gefäßbiologie, Immunologie und Entzündung, Lehrstuhl für Pharmakologie, Medizinische Universität Graz

Ursprünglich erschienen:
UIM 09|2019
UIM 09|2019
