


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Pathogenetische Grundlagen der Autoimmunthyreoiditis – Genetische Prädisposition und Umweltfaktoren
14. September 2012
Bei der Entwicklung einer Autoimmunthyreoiditis wirken wie bei den meisten Autoimmunerkrankungen (angeborene) genetische Veranlagungen mit (erworbenen) Umweltfaktoren zusammen (siehe Abb. 1, nach Saranac 2012). Die genetische Prädisposition besteht in einer allgemeinen Veranlagung für die Entwicklung von Autoimmunerkrankungen sowie in organspezifischen Faktoren. Die allgemeine Veranlagung erklärt auch das häufigere Vorkommen verschiedener (endokriner) Autoimmunerkrankungen bei Patienten mit Autoimmunthyreoiditis (Vitiligo, Morbus Addison, Hypoparathyreoidismus, Typ-1-Diabetes, Autoimmungastritis).
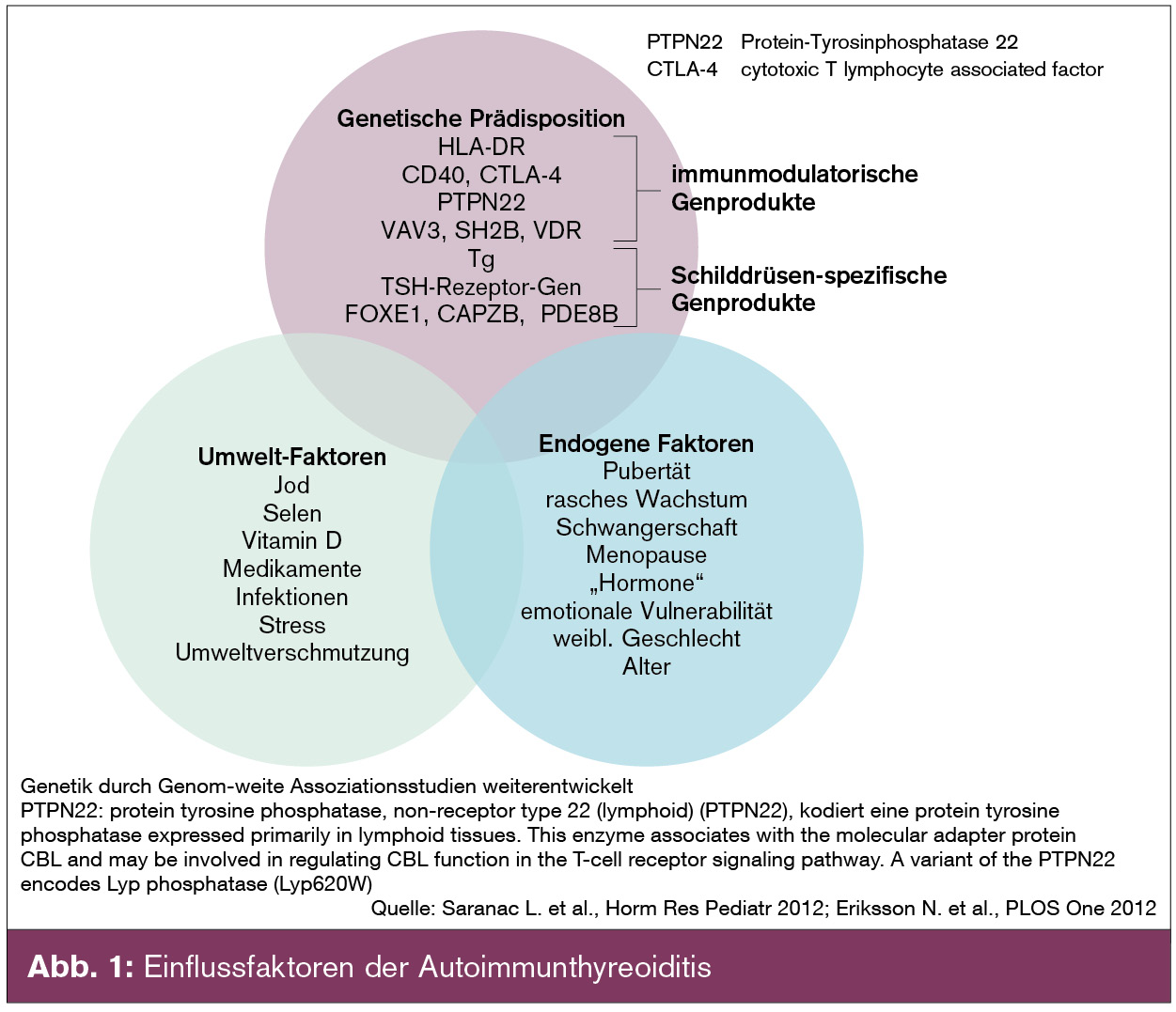
Genetische Faktoren
Zur Entstehung einer Autoimmunerkrankung müssen autoreaktive T-Zellen durch die normalen Auslesemechanismen (Deletion und Suppression) rutschen. Bei einer seltenen monogenetischen, autosomal-rezessiv vererbten Form (endokrine Polyendokrinopathie Typ 1 oder APECED, OMIM 240300, Mutation im Transkriptionsfakor AIRE) ist durch die Mutation im AIRE-Gen die Expression von Autoantigenen im Thymus gestört, sodass die negative Selektion autoreaktiver T-Zellen unterbleibt. Neben anderen Problemen gehört auch eine Autoimmunthyreoiditis zum Phänotyp. Eine minimale Zahl autoreaktiver T-Zellen übersteht jedoch immer diesen Ausleseprozess und wird durch weitere Mechanismen in Schach gehalten.
Bei der T-Zell-Aktivierung durch Antigen-präsentierende Zellen (APC) erkennt die T-Zelle über den T-Zell-Rezeptor das durch MHC-Klasse-II-Antigene gebundene Peptidantigen. Unterschiedliche MHC-Klasse-II-Antigene können manche Autoantigene besser präsentieren und prädisponieren daher für manche Autoimmunerkrankungen (bzw. wirken protektiv). CD80- und CD86-Moleküle auf APCs dienen als Kostimulatoren und interagieren mit CD28, dem primären kostimulatorischen Molekül auf T-Zellen, um die Aktivierung zu verstärken. An der T-Zell-Oberfläche vorhandenes zytotoxisches T-Lymphozyten-Antigen 4 (CTLA-4, CD152) bindet ebenfalls an CD80/CD86 auf APC, wodurch allerdings die Aktivierungsschwelle erhöht statt vermindert wird.
Der CT60-G/G-Phänotyp ist bei Autoimmunthyreoiditis häufiger, er führt zu geringerer Expression auf T-Zellen und geringerer Inhibition der Immunantwort bzw. eine Hemmung dieser CTLA-4-Funktion ist auch durch monoklonale Antikörper möglich. Dies führt zu verstärkten (Auto-)Immunreaktionen und wird als Therapie bei metastasierten Melanomen durch die verstärkte Anti-Tumor-Immunität genutzt (Ipilimumab, Yervoy®). Bei medikamentöser CTLA-4-Hemmung treten sehr häufig Endokrinopathien inkl. Autoimmunthyreoiditiden auf.
Phosphatase LYP: Nach der (durch mit dem T-Zell-Rezeptor assoziiertes CD3 vermittelten) Signaltransduktion ist in der Signalkaskade die Phosphatase LYP (kodiert durch das PTPN22-Gen) zwischengeschaltet, über die eine Dämpfung erfolgt. Bestimmte Polymorphismen führen zu einem stärkeren LYP-Abbau und so zu einer stärkeren T-Zell-Aktivierung und häufigeren Autoimmunerkrankungen.
Der Transkriptionsfaktor FoxP3 dirigiert die Entwicklung (Differenzierung) und Funktion regulatorischer T-Zellen (Tregs) die für die Immuntoleranz (nicht eliminierter) autoreaktiver T-Zellen sorgen können. X-chromosomal rezessiv vererbte Loss-of-Function-Mutationen im FOXP3-Gen führen beim Knaben zum IPEX-Syndrom (OMIM 304790) (für Immundysregulation, Polyendokrinopathie, Enteropathie, X-linked). Als endokrines Leiden tritt bei dieser seltenen Erkrankung neben neonatalem/im ersten Lebensjahr auftretendem Typ-1-Diabetes eine Thyreoiditis auf, andere Endokrinopathien sind sehr selten.
Diagnostische Parameter
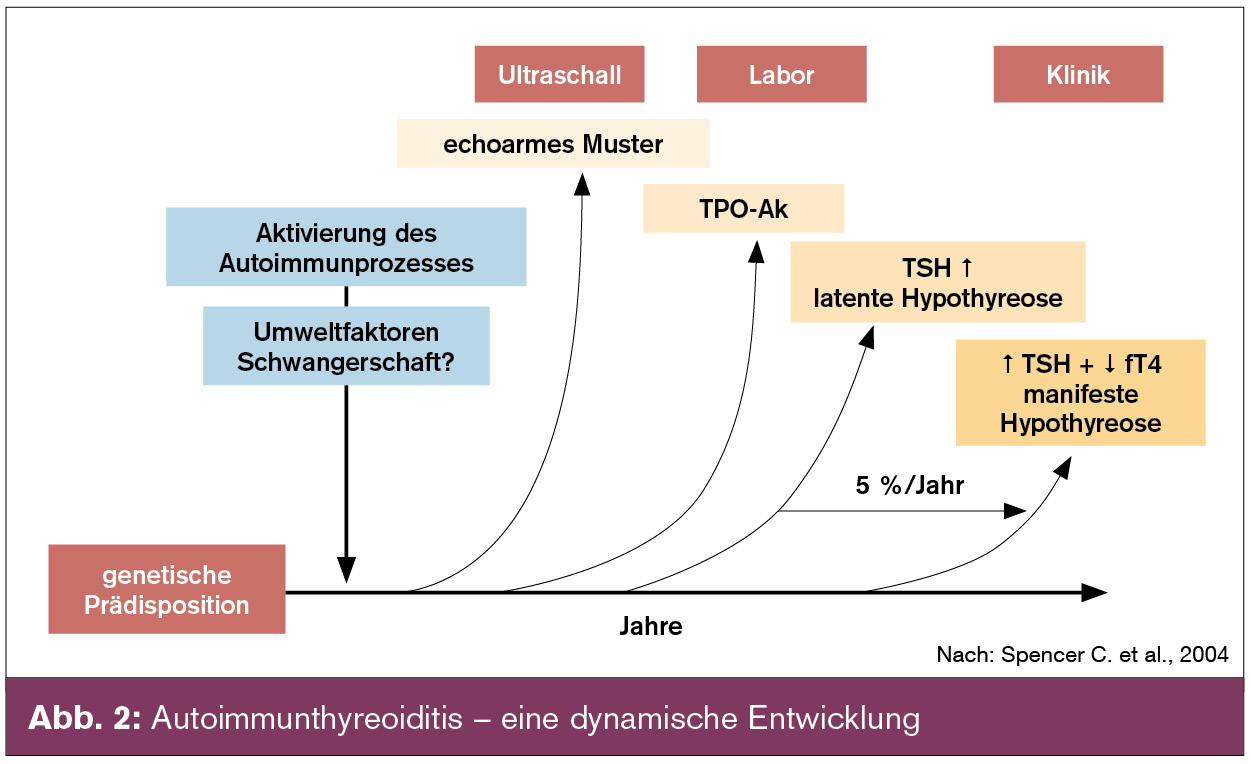
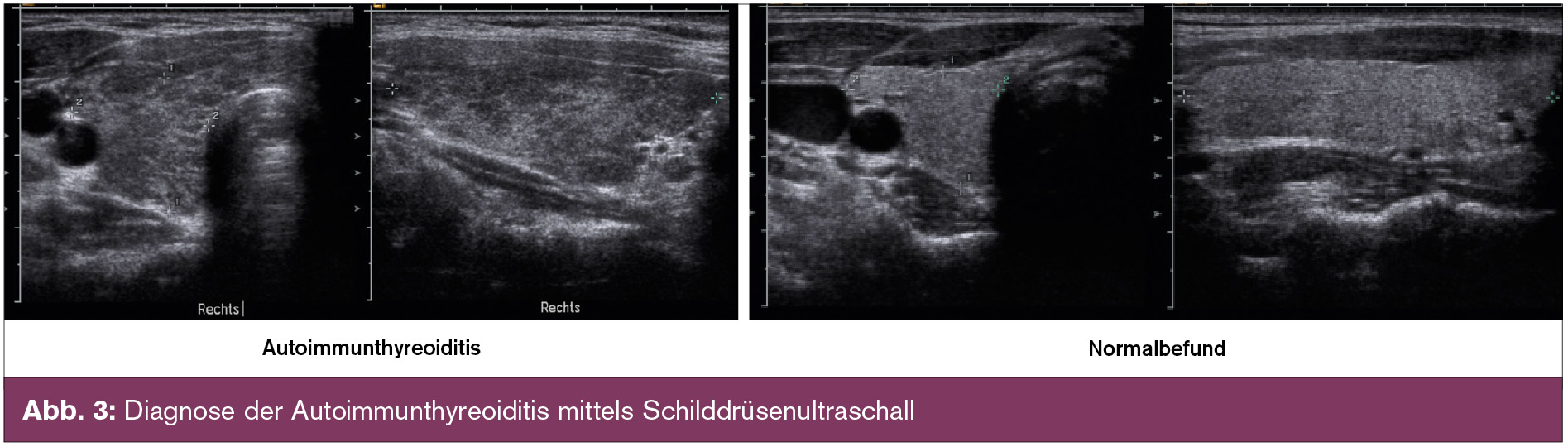
Im klinischen Verlauf der Autoimmunthyreoiditis treten früh Schilddrüsen-Autoantikörper auf (Abb. 2). Ein Nachweis von Thyreoperoxidase-(TPO)-Antikörpern (eingeschränkt Thyreoglobulin-[Tg]-Antikörper) spricht für die Entwicklung einer Autoimmunthyreoiditis und für das Risiko einer zukünftigen Hypothyreose. Pathophysiologisch sind sie Marker ohne kausale Bedeutung. Wiederholte Bestimmungen dieser Antikörper sind von fragwürdigem klinischem Nutzen und führen aufgrund der natürlichen Titerschwankungen lediglich zur Neurotisierung von Patienten. Nicht vorhandene Autoantikörper schließen eine Autoimmunthyreoiditis nicht aus, dieser Antikörper-negative „jugendliche“ Phänotyp, zu dem auch das frühere Manifestationsalter, die geringere weibliche Präponderanz, die nur latente Funktionsstörung sowie die meist nicht vergrößerte Schilddrüse gehört, löst zunehmend die klassische Hashimoto-Thyreoiditis ab (Benvenga S. et al., 2008). Die Schilddrüsensonografie stellt besonders in Antikörper-negativen Fällen ein exzellentes diagnostisches Werkzeug dar, da sehr häufig das typische sonografische Bild der Autoimmunthyreoiditis nachgewiesen werden kann (Abb. 3).
In diesem Zusammenhang sei auch festgehalten, dass die Dichotomie in atrophe und hypertrophe (= eigentliche Hashimoto-Thyreoiditis) Form der Autoimmunthyreoiditis eine lang gehegte Chimäre darstellt (Publikationen zeigten unterschiedliche HLA-Assoziationen etc.). Eigene Daten wie auch eine gut durchgeführte dänische Untersuchung zeigt eindeutig eine Normalverteilung des Schilddrüsenvolumens bei der Autoimmunthyreoiditis. Auch ist die atrophe Form keine „ausgebrannte“ Thyreoiditis-Spätform.
Der Grund für das Zunehmen (v. a. der „jugendlichen“ Form) der Autoimmunthyreoiditis ist unklar, ist aber durch einen „lead time bias“ (früheres Erkennen durch häufigere und frühere Laboruntersuchungen) allein nicht erklärbar. Hinweise deuten auf eine mögliche Rolle eines Vitamin-D-Mangels (u. a. durch zunehmendes Problembewusstsein bezüglich Sonnenlichtexposition), einen Selenmangel, die bessere Jodversorgung sowie Umweltgifte u. a. aus der petrochemischen Industrie (wie z. B. Trichloräthylen). Obwohl die vielfältigen Wirkungen von Vitamin D auf das angeborene und adaptive Immunsystem sowie Querschnittuntersuchungen eine Rolle des Vitamin-D-Mangels bei der Autoimmunthyreoiditis nahelegen, zeigt eine rezente prospektive Untersuchung keinen Zusammenhang. Ebenso ist der Selenmangel als Auslöser nicht gesichert, noch weniger ist eine blinde Therapie mit Selen gegenwärtig zu empfehlen.
Literatur beim Verfasser

Ursprünglich erschienen:
UIM 07|2012
UIM 07|2012
Herausgeber: Univ.-Prof. Dr. Günter J. Krejs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2012-09-14
Zur Ausgabe »
Publikationsdatum: 2012-09-14
Zur Ausgabe »