


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Immunadrenopathien – State of the Art
14. September 2012
Die menschliche Nebennierenrinde (NNR) ist in drei Zonen unterteilt: Zona glomerulosa (Mineralokortikoid-Synthese), Zona fasciculata (Glukokortikoid-Synthese) und Zona reticularis (Androgen-Synthese). Die Immunadrenopathien führen praktisch immer zu einer Zerstörung aller drei Zonen (primäre NNR-Insuffizienz = Morbus Addison) und sind von der sekundären, hypophysären NNR-Insuffizienz abzugrenzen, wo nur die ACTH-regulierten Zonae fasciculata und reticularis ausfallen.
Epidemiologie, Pathophysiologie
Die primäre NNR-Insuffizienz ist eine seltene Erkrankung (Prävalenz: ca. 100 Erkrankte pro Million Einwohner in Europa). Als Folge der modernen tuberkulostatischen Therapie ist heute die Autoimmunadrenalitis mit 80–90 % die bei weitem häufigste Krankheitsursache. Bei Patienten mit Autoimmunadrenalitis findet man im Blut zirkulierende Antikörper gegen NNR-Antigene, wobei insbesondere die Antikörper gegen die 21-Hydroxylase (21-OH-Ab) eine relativ hohe Sensitivität und Spezifität für die Autoimmunadrenalitis haben. 40–50 % der Patienten mit Autoimmunadrenalitis erkranken im Rahmen einer polyglandulären Insuffizienz Typ 1 oder Typ 2 (Autoimmune Polyendocrinopathy Type 1, Type 2 = APS-1, APS-2) an weiteren Autoimmunerkrankungen.
APS-1: Das autoimmun-pluriglanduläre Syndrom 1 (APS-1) oder APECED-Syndrom (Autoimmune Polyendocrinopathy Candidiasis Ectodermal Dystrophy Syndrome) ist wesentlich seltener als das APS-2 und umfasst mindestens 2 der 3 folgenden Krankheitsentitäten: 1. primäre NNR-Insuffizienz, 2. Hypoparathyreoidismus, 3. chronische mukokutane Candidiasis. Die autosomal-rezessiv vererbte Erkrankung manifestiert sich zumeist in Kindheit und früher Jugend, wobei meistens einer neonatal schon vorhandenen Candidiasis der Hypoparathyreoidismus (3.–10. Lebensjahr) und dann die primäre NNR-Insuffizienz (11.–15. Lebensjahr) folgt. Das APS-1 ist eine monogenetische Autoimmunerkrankung, die durch Mutationen im Bereich des AIRE-1-Gens (Autoimmune Regulator 1) verursacht wird.
APS-2: Beim wesentlich häufigeren autoimmun-pluriglandulären Syndrom 2 (APS-2, Schmidt-Syndrom) findet man neben einer primären NNR-Insuffizienz eine Autoimmunthyreopathie (meist Hashimoto-Thyreoiditis, aber auch Morbus Basedow), seltener einen Diabetes mellitus Typ 1, eine primäre Ovarialinsuffizienz oder einen Vitamin-B12-Mangel. Das Manifestationsalter liegt zwischen dem 30. und 50. Lebensjahr, Frauen werden bevorzugt betroffen. Im Gegensatz zu den Patienten mit APS-1 findet man beim APS-2 eine Assoziation zum HLA-System (HLA-DR3 und -DR4). Die genetische Ursache des APS-2 ist nicht geklärt; eine komplexe polygenetische Ätiologie ist sehr wahrscheinlich.
Klinisches Bild
Die klinische Symptomatik der Patienten mit NNR-Insuffizienz lässt sich weitgehend aus dem Ausfall der entsprechenden Hormone ableiten (Tab., Abb.).
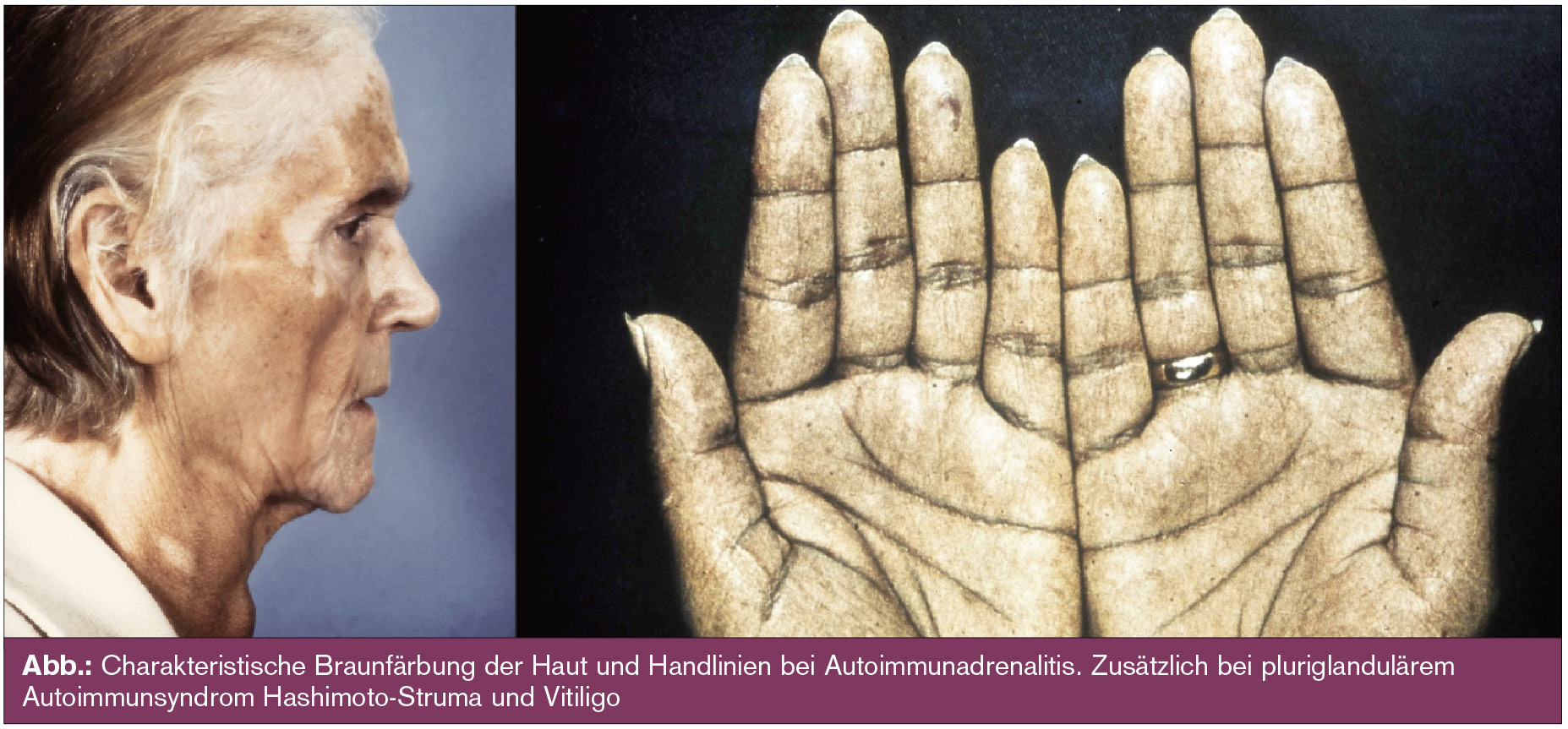
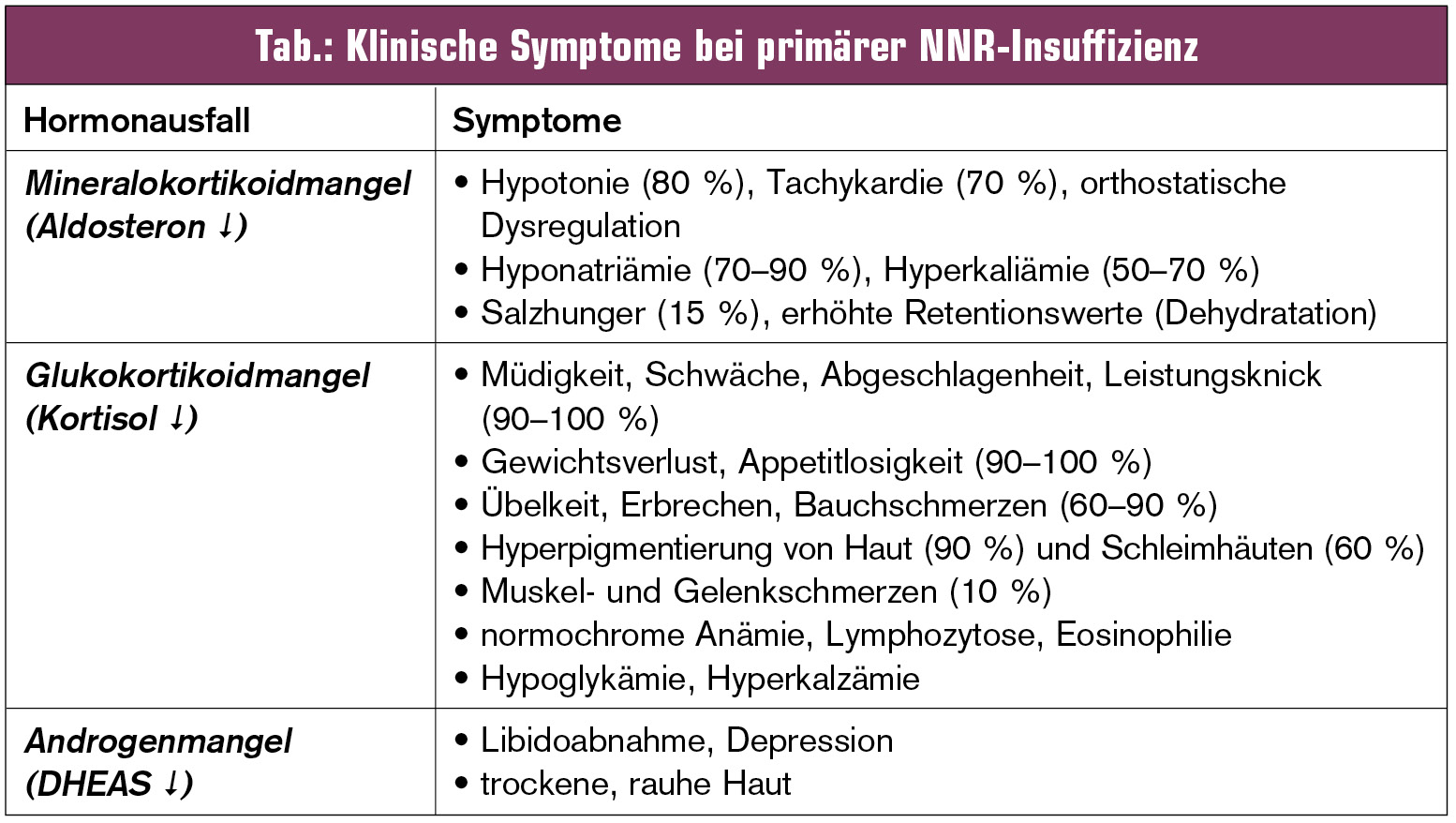
Diagnostik
Als erster diagnostischer Test kann eine morgendliche Blutabnahme für Serum-Kortisol bereits die Diagnose sichern (S-Kortisol < 36 ng/ml [ 180 ng/ml [> 500 nmol/l]). Ein parallel bestimmtes Plasma-ACTH erlaubt die Differenzierung zwischen primärer (ACTH erhöht) und sekundärer Insuffizienz (ACTH normal oder erniedrigt).
Bei diesem Vorgehen muss man einen relativ großen Teil der Patienten (S-Kortisol zwischen 36 und 180 ng/ml) weiter untersuchen, so dass bei Verfügbarkeit die direkte, initiale Durchführung eines ACTH-Kurztests sinnvoll ist.
Der ACTH-Kurztest hat für die primäre NNR-Insuffizienz mit einem Cut-off-Wert von 180 ng/ml eine sehr gute Sensitivität. Ein Anstieg des Kortisols auf > 180 ng/ml schließt damit eine primäre NNR-Insuffizienz 100%ig aus. Bei der sekundären NNR-Insuffizienz, bei der nur indirekt die Atrophie und die ACTH-Rezeptor-Down-Regulation der zona fasciculata aufgrund eines länger bestehenden ACTH-Mangels zu einem pathologischen ACTH-Test führt, hat der ACTH-Kurztest eine etwas geringere Sensitivität. Daher muss man hier in einem Graubereich zwischen 180 und 220 ng/ml (500–600 nmol/l) unbedingt noch weitere Tests wie den Insulin-Hypoglykämie- oder Metopiron®-Test durchführen.
In der weiterführenden Diagnostik bestimmt man zur Beurteilung der Zona glomerulosa das Plasma-Aldosteron und die Plasma-Reninkonzentration und zur Beurteilung der Zona reticularis das Dehydroepiandrosteron-Sulfat (DHEAS) im Serum.
Therapie
Eine Heilung der Immunadrenopathien durch z. B. immunsuppressive Therapie ist nicht bekannt. Alle Patienten benötigen als Glukokortikoid-(GC)-Substitution Hydrokortison, Kortisonazetat (wird in der Leber zu Hydrokortison metabolisiert) oder auch Prednisolon. Die Kortisolsekretionsrate beim Menschen beträgt 9–11 mg/m2 Körperoberfläche, was einem durchschnittlichen Substitutionsbedarf von 15–20 mg Hydrokortison täglich entspricht. Die meisten Therapeuten geben die GC in 2 oder 3 Tagesdosen, um die zirkadiane Rhythmik der Kortisol-Sekretion nachzuahmen. Da die derzeit verfügbaren Substanzen zur GC-Substitution die Tagesrhythmik nur schlecht nachahmen und sich NNR-insuffiziente Patienten in größeren Vergleichsstudien schlechter belastbar fühlen, sind retardierte Hydrokortison-Tabletten in Entwicklung (Plenadren®, Chronocort®). Bei erhöhtem Stress wie z. B. fieberhaften Erkrankungen sollte die Tagesdosis verdoppelt oder verdreifacht werden. Kann Hydrokortison im Rahmen eines gastrointestinalen Infekts nicht oral eingenommen werden, sollte jeder Patient Hydrokortison- oder Prednisolon-Suppositorien in seiner Notfallapotheke haben. Treten zusätzlich Diarrhöen auf, muss der Patient umgehend einen Arzt oder ein Krankenhaus zur parenteralen Hydrokortisonzufuhr aufsuchen. Jeder Patient mit NNR-Insuffizienz muss einen GC-Notfallausweis besitzen und regelmäßig – möglichst unter Einbeziehung der Angehörigen – geschult werden. Die Kontrolle der GC-Dosierung erfolgt klinisch, da es diesbezüglich keine geeigneten Laborparameter gibt.
Die Mineralokortikoid-Substitution erfolgt durch die orale Gabe von 0,05–0,2 mg 9a-Fluoro-Hydrokortison (Astonin® H) pro Tag. Als klinische bzw. laborchemische Kontrollgrößen für die richtige Dosierung sind Körpergewicht, Blutdruck, Herzfrequenz und Serum-Kalium geeignet. Genauer ist die Messung der Plasma-Reninkonzentration, welche in den oberen Normbereich gesenkt werden soll.
Eine Androgensubstitution mit 25–50 mg Dehydroepiandrosteron (DHEA) sollte bei Patienten mit entsprechenden Symptomen (Tab.) in Erwägung gezogen werden. Da diese Substanz nicht als Medikament zugelassen ist, können sich die Patienten dieses über Internetanbieter oder über spezialisierte Apotheken besorgen (nur auf Privatrezept). Sollte es hierunter nach 3 bis 6 Monaten zu keiner subjektiven Besserung im Befinden kommen, muss diese Substitution nicht fortgeführt werden.
RESÜMEE: Immunadrenopathien sind seltene, durch den Glukokortikoidmangel potenziell lebensbedrohliche Erkrankungen, die trotz eines charakteristischen Krankheitsbildes oft spät erkannt werden. Sowohl die biochemische Diagnostik mittels ACTH-Kurztest als auch die Substitutionstherapie mit Hydrokortison sind einfach und effektiv. Für den Patienten ist hiermit oft ein langer Leidensweg mit geringem Aufwand zu beenden. In der Langzeitbetreuung sind Notfallausweis und -Schulung zur Vermeidung von Addison-Krisen und das Beachten möglicher assoziierter Autoimmunerkrankungen (Hashimoto, DM-1, Vitamin-B12-Mangel) von Bedeutung.

Ursprünglich erschienen:
UIM 07|2012
UIM 07|2012
Herausgeber: Univ.-Prof. Dr. Günter J. Krejs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2012-09-14
Zur Ausgabe »
Publikationsdatum: 2012-09-14
Zur Ausgabe »