


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Plasmapherese und Immunadsorption
22. April 2020
Die Plasmapherese und die Immunadsorption (IA), zwei breit eingesetzte Apheresetechniken, sind medizinische Verfahren, deren therapeutische Wirkung auf der Elimination bestimmter krankheitserregender Bestandteile im Blut basiert.1
Bei der Plasmapherese bzw. dem therapeutischen Plasmaaustausch (TPA) wird das Plasma von zellulären Blutkomponenten getrennt und letztendlich entfernt bzw. durch Substitutionslösungen (z. B. Humanalbumin oder Fresh Frozen Plasma), oft in Kombination, ersetzt. Darüber hinaus ist es möglich, für den Körper bedeutsame Faktoren durch die Plasmarückgabe zuzuführen, beispielsweise Komplementfaktor H beim hämolytisch-urämischen Syndrom. Dies ist vor allem in großen Mengen nur dann möglich, wenn man den gleichen Teil an Faktor-H-Autoantikörpern entfernt.2
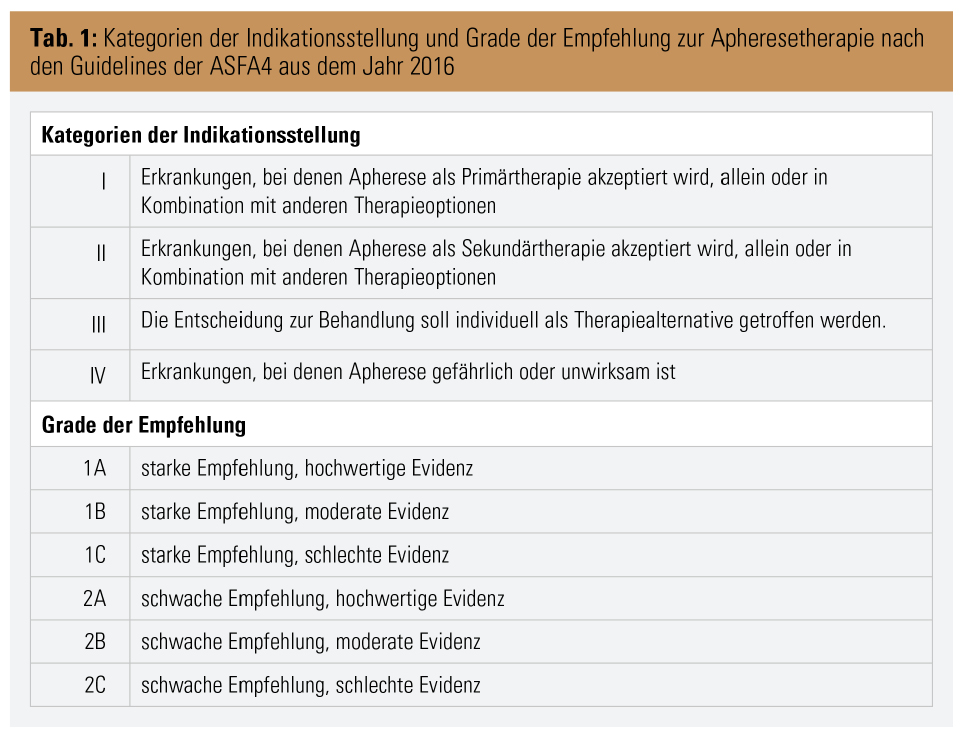
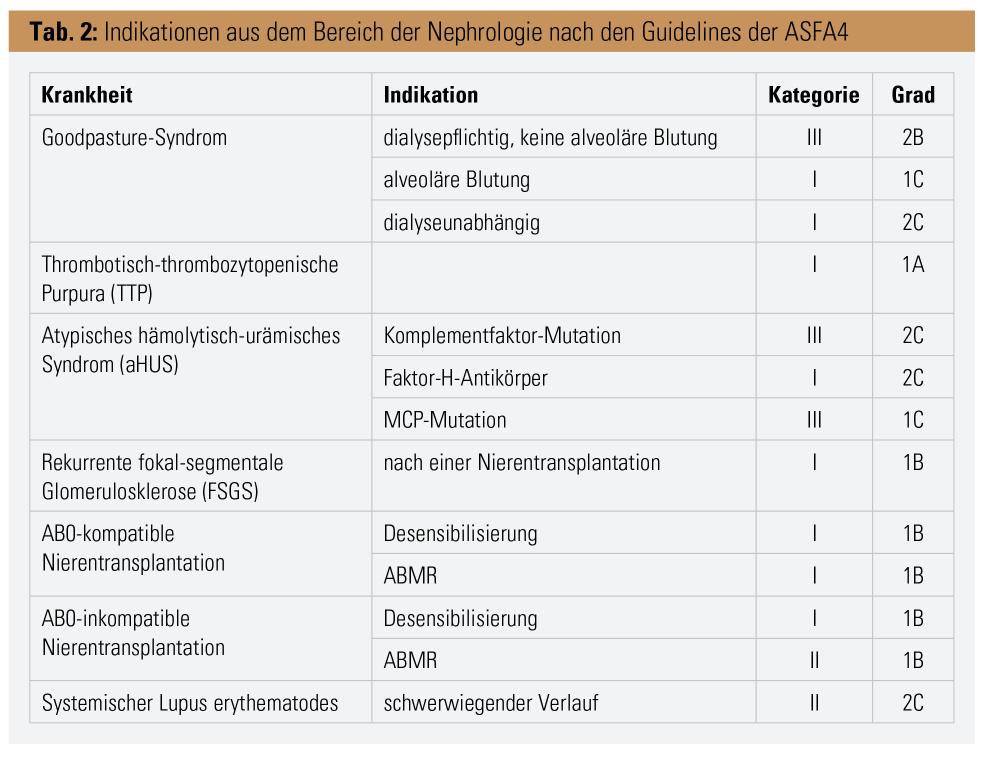
Bei der Immunadsorption (IA) handelt es sich um eine Technik, bei der bestimmte Bestandteile, vor allem Immunglobuline (Ig), selektiv aus dem Blutkreislauf entfernt werden können. Dies geschieht durch Verwendung sog. Adsorber, welche über matrixgebundene spezifische Liganden Ig (vor allem IgG) spezifisch binden.3 Beide Verfahren konnten sich in einigen Teilgebieten der Medizin als erfolgreiche Therapieansätze unter Beweis stellen, zusammengefasst in den Guidelines der American Society of Apheresis (ASFA), eingeteilt in 4 Kategorien der Indikationsstellung mit unterschiedlichen Graden der Empfehlung (Tab. 1).4 Aus dem Bereich der Nephrologie betrifft das im Wesentlichen die in der Tabelle 2 gelisteten Krankheitsbilder.4
Nephrologische Einsatzgebiete
Das Goodpasture-Syndrom beschreibt eine sehr seltene Autoimmunerkrankung mit renaler und pulmonaler Beteiligung. Laborchemisch lassen sich hier zirkulierende Antikörper gegen glomeruläre Basalmembran (GBM) feststellen, welche die nichtkollagene Domäne der α3-Kette vom Typ-IV-Kollagen binden. Diese Art von Kollagen ist ein wichtiger Bestandteil der Basalmembran von Lunge und Niere, folglich kann es hier zu schweren organischen Schäden kommen. Renal manifestiert sich die Krankheit durch ein gelegentlich dialysepflichtiges akutes Nierenversagen (Bild einer rapid progredienten Glomerulonephritis). Mögliche pulmonale Symptome sind Hämoptysen und respiratorische Insuffizienz. Die Therapie der Wahl besteht hier in einer Kombination aus TPA und Immunsuppressiva, meist kommt es hierunter innerhalb von 2 Wochen zu einem Abfallen der Anti-GBM-Antikörper unter die Nachweisgrenze.4 Auch die Anwendung der IA kann zu einer effizienten Eliminierung von Anti-GBM-Antikörpern führen und ist somit dem TPA in der Wirkungsweise nicht unterlegen, vor allem bei Patienten, die bereits an einem dialysepflichtigen akuten Nierenversagen leiden.5
Fokal segmentale Glomerulosklerose (FSGS): Bei der FSGS handelt es sich um eine Form der Glomerulonephritis, deren Vorliegen anhand einer Nierenbiopsie nachgewiesen wird. Ein charakteristischer histologischer Befund weist hier fokale Bereiche von Sklerosierung und hyalinen Einlagerungen einiger Glomeruli neben anderen völlig intakten Glomeruli auf. Erkrankte präsentieren sich klinisch mit einer ausgeprägten Proteinurie, Hypertonie und Ödembildung. 80 % der FSGS-Fälle sind idiopathisch, andere Ursachen sind z. B. Mutationen in bestimmten Podozytengenen, Medikamente und Drogen (Heroin, Lithium) sowie Hypertonie oder bei Einnierigkeit bzw. Zustand nach Nephrektomie. Als Auslöser der idiopathischen FSGS vermutet man einen unidentifizierten Plasmafaktor, welcher die Filtrationsbarriere verletzt und die glomeruläre Permeabilität erhöht. Unterstützt wird diese Theorie dadurch, dass eine FSGS in einer transplantierten Niere erneut auftreten kann.
Ein Wiederauftreten ist einige Stunden bis 2 Jahre nach der Transplantation möglich. Unbehandelt führt eine rekurrente FSGS letztendlich zum permanenten Transplantatverlust innerhalb von Monaten. Patienten, die Transplantate durch ein Rezidiv verloren haben, haben ein > 80%iges Risiko für ein erneutes Auftreten von FSGS infolge weiterer transplantierter Nieren. Auch wenn der TPA bei Patienten mit primärer FSGS der Eigennieren zu keiner klinischen Verbesserung führt, hilft er überraschenderweise bei Nierentransplantierten mit FSGS-Rezidiv in Kombination mit Kortison und anderen Immunsuppressiva.4, 6, 7
AB0-kompatible und -inkompatible Nierentransplantation: Als Reaktion auf zunehmenden Organmangel und erhöhte Sensibilisierung bei Organempfängern werden immer mehr Transplantationen von immunologisch inkompatiblen Nieren durchgeführt. HLA-Antikörper, entstanden durch frühere Einwirkung von Schwangerschaften, Bluttransfusionen oder Transplantationen, können gegen ein donorspezifisches Antigen (DSA) gerichtet sein und hyperakute, akute oder chronische antikörpervermittelte Abstoßungsreaktionen (Antibody-mediated Rejection – ABMR) verursachen. Darüber hinaus müssen Patienten mit erhöhtem Screening auf HLA-Antikörper (Panel-reactive Antibodies – PRA) öfters signifikant länger auf der Transplantationsliste verbleiben, da es schwieriger ist, einen HLA-kompatiblen Spender zu finden. Infolgedessen schreibt man hier dem TPA und der IA eine immer größer werdende Bedeutung zu, vor allem, um bereits vorhandene Antikörper-Titer zu senken und so durch Desensibilisierung die Möglichkeiten zur Transplantation auszubauen. Zusätzlich werden aufgrund von Organknappheit AB0-inkompatible lebende Spender herangezogen. In diesen Fällen betrifft die Unverträglichkeit das natürliche Vorkommen von Antikörpern im Empfänger gegen das A- oder/und B-Blutgruppenantigen des Spenders. Wesentliche AB0-Inkompatibilität kommt in ca. 35 % zufälliger Spender-Empfänger-Konstellationen vor.4, 8–10
Systemischer Lupus erythematodes (SLE) beschreibt eine chronische, mehrere Organsysteme betreffende, entzündliche Erkrankung autoimmunologischen Ursprungs. Von der Krankheit sind vorwiegend Frauen im gebärfähigen Alter betroffen (ca. 90 %). Klinische Symptome sind weitgehend unspezifisch und reichen von Abgeschlagenheit, Hauterscheinungen, Gelenk- und Muskelschmerzen über Anorexie und Gewichtsverlust bis hin zu migräneähnlichen Kopfschmerzen und Fieber und können auf die Beteiligung eines oder mehrerer Organsysteme zurückgeführt werden. Der Verlauf der Erkrankung ist chronisch rezidivierend sowie remittierend, Ausmaß und Geschwindigkeit des Fortschreitens sind aber variabel.4 Autoantikörper sind kennzeichnend für diese Erkrankung und tragen wesentlich zur Pathogenese bei. Sie binden direkt oder über Immunkomplexe an Zellen und Gewebe und leiten eine Aktivierung des Komplementsystems sowie Entzündungsprozesse ein und verursachen Gewebeschädigungen.11 Erhöhte systemische Krankheitsaktivität kann durch die Anwendung von IA vermindert werden.12
Thrombotische Mikroangiopathien (TMA): Die thrombotisch-thrombozytopenische Purpura (TTP) gehört als Krankheitsbild zu den sogenannten TMA. Ihr zugrunde liegt der schwere Mangel (< 10 %) an Plasma-ADAMTS13-Enzymaktivität, durch deren Ausbleiben der Von-Willebrand-Faktor (vWF) im Blut nicht gespalten werden kann und somit die Quervernetzung der Thrombozyten sowie deren Adhärenz an verletzte Gefäßwände verhindert wird. Folglich kommt es zur Entstehung von Thromben in kleinsten Gefäßen, bestehend aus vWF-Multimeren, da deren Normalverteilung nicht mehr aufrechterhalten werden kann. Dies führt zu einer organischen Minderdurchblutung durch mechanische Destruktion von Erythrozyten, vor allem in der Niere und im Gehirn. In 90 % der Fälle hat die Krankheit unbehandelt den Tod zur Folge. TPA konnte die Mortalität der idiopathischen TTP auf unter 10 % senken und den klinischen Ausgang erheblich verbessern. Er sollte unverzüglich nach Feststellung der Erkrankung begonnen werden.4, 13
Das zweite bedeutende Krankheitsbild aus der Gruppe der TMA ist das hämolytisch-urämische Syndrom (HUS), geprägt durch Thrombozytopenie, hämolytische Anämie durch mikroangiopathische Schädigung von kleinen Blutgefäßen sowie akutes Nierenversagen bis hin zur Dialysepflicht, oft binnen kurzer Zeit. Während der typische Auslöser Shiga-Toxin-produzierende E.-coli-Stämme sind, verlaufen 5–10 % der Fälle atypisch. Hier manifestiert sich eine Überaktivierung des Komplementsystems aufgrund des Fehlens von essenziellen regulierenden Proteinen. Als Therapie der Wahl gilt hier ebenso der TPA, mutierte Komplementfaktoren können erfolgreich aus dem Plasma entfernt und durch funktionelle Faktoren im Plasmaaustausch ersetzt werden.4, 14
Zusammenfassung
In einigen Teilgebieten der Medizin haben sich die Immunadsorption und der Plasmaaustausch als umfassend einsetzbare Therapiemethoden etabliert. Vor allem im Bereich der Erkrankungen mit autoimmunologischer Pathogenese schreibt man den Apheresetechniken eine wertvolle Bedeutung zu, oft auch in Kombination mit zusätzlichen immunsuppressiven Therapien. Im Bereich der Organtransplantationen kommt es aufgrund von Mangel und zunehmender Sensibilisierung bei Empfängern zunehmend zu immunologischer Inkompatibilität, Immunadsorption und Plasmaaustausch ermöglichen durch Desensibilisierung und Antikörper-Titersenkung einer Vielzahl von Organempfängern die Verkürzung der Wartedauer auf ein Organ und die Vermeidung von Abstoßungsreaktionen nach Transplantation.
1 Visvardis G et al., Renal Failure 2004; 26(5):569–574
2 Tölle M, Dialyse aktuell 2015; 19(8):424–436
3 Bennani HN et al., Case reports in nephrology 2019; Article ID 7304786, 4 pages
4 Schwartz J et al., Journal of Clinical Apheresis 2016; 31:149–338
5 Biesenbach P et al., PLoS ONE 2014; 9(7):e103658
6 Martin-Moreno PL et al., Blood Purif 2018; 46:90–93
7 Kandus A et al., Therapeutic Apheresis and Dialysis 2013; 17(4):438–443
8 Gubensek J et al., Therapeutic Apheresis and Dialysis 2016; 20(3):240–245
9 Fehr T et al., Transplant International 2012; 25:623–632
10 Rostaing L et al., Kidney International 2014; 84:232–247
11 Biesenbach P et al., Atherosclerosis Supplements 2009; 10:114–121
12 Kronbichler A et al., Autoimmunity Reviews 2016; 15:38–49
13 Thurman JM et al., Clin J Am Soc Nephrol 2018; 13:933–936
14 Kaplan BS, Ruebner RL, Spinale JM, Copelovitch L, Current treatment of atypical hemolytic uremic syndrom. Intractable & Rare Diseases Research 2014; 3(2):34–45

AutorIn: Dr. Thomas Berndl
Klinische Abteilung für Nephrologie und Dialyse, Universitätsklinik für Innere Medizin III, Medizinische Universität Wien

Ursprünglich erschienen:
UIM 03|2020
UIM 03|2020
