


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Zielgerichtete Therapien bei Weichteil- und Knochensarkomen
13. Juli 2012
Aufgrund ihrer Inzidenz von nur etwa 6 Neuerkrankungen pro 100.000 Einwohner und Jahr werden maligne mesenchymale Neoplasien häufig als eine Krankheitsentität – Sarkome – angeführt. Tatsächlich unterscheiden sich die über 60 histologischen Subtypen erheblich hinsichtlich der zugrunde liegenden molekularen Veränderungen und in ihrem klinischen Verhalten.
Letzteres reicht von unbehandelt äußerst aggressiv verlaufenden, chemotherapiesensitiven Tumoren (z. B. Ewing- Sarkom, embryonales Rhabdomyosarkom) bis hin zu über lange Zeit indolent verlaufenden, chemo- und strahlentherapierefraktären Tumoren (z. B. hochdifferenzierte Liposarkome oder konventionelle Chondrosarkome). Aus diesen Gründen erfolgen die bildgebende und (molekular)pathologische Diagnostik sowie die interdisziplinäre Therapieplanung idealerweise individualisiert an Zentren mit entsprechenden Fallzahlen.
Molekulargenetische Veränderungen als Target zielgerichteter Therapien
Direkte Targets: Prinzipiell lassen sich Sarkome mit einfachen, meist pathognomischen genetischen Aberrationen von solchen mit komplexen und interindividuell heterogenen Karyotypen unterscheiden. Erstere sind seltener und umfassen beispielsweise aktivierende Punktmutationen oder kleine Insertionen/Deletionen in einem oder wenigen Genen. Dazu zählen z. B. gastrointestinale Stromatumoren (GIST, siehe Beitrag von Prof. Thomas Brodowicz) mit ihren prototypischen Mutationen der c-Kitund/ oder PDGFRA-Rezeptortyrosinkinasen. Deren Aktivität ist für die Aufrechterhaltung des malignen Wachstums von GIST essenziell und darüber hinaus einer Therapie mit Tyrosinkinasehemmern wie Imatinib oder Sunitinib gut zugänglich. In ähnlicher Weise spielen Mutationen, die zu einer erhöhten Aktivität der Indian-Hedgehog-Signalkaskade führen, bei konventionellen Chondrosarkomen eine zentrale Rolle. Demensprechend finden sich speziell für die Therapie dieser chemotherapieresistenten Sarkome Smoothened-Inhibito – ren in klinischer Erprobung.
Andere einfache molekulare Veränderungen umfassen chromosomale Aberrationen, die zu Genfusionen und konsekutiver Expression entsprechender Fusionstranskripte führen. Diese können ebenfalls Kinasen als Translokationspartner inkludieren (z. B. COL1A1- PDGFRB bei Dermatofibrosarcoma protuberans, DFSP) und so einer zielgerichteten Therapie gut zugänglich sein (z. B. Imatinib bei DFSP).
Indirekte Targets: Häufiger jedoch inkludieren diese Fusionsgene chimäre Transkriptionsfaktoren (z. B. EWSR1-FLI1 bei Ewing-Sarkomen), welche sich aufgrund ihrer intrazellulären Lage und dem Fehlen einer hemmbaren katalytischen Enzymaktivität bislang einem pharmakologischen targeting entzogen haben. Neue Ansätze verfolgen jedoch die zielgerichtete Therapie jener von diesen Transkripten hochregulierten Signalkaskaden. Beispielsweise führt das EWSR1-ATF1-Fusionstranskript bei Klarzellsarkomen über Aktivierung des Transkriptionsfaktors MITF zur vermehrten Expression und Aktivierung der MET-Kinase. Aufgrund vielversprechen – der präklinischer und präliminärer Daten an kleinen PatientInnenkollektiven wird derzeit im Rahmen einer internationalen Studie der potenzielle Nutzen des kombinierten ALK/MET-Inhibitors Crizotinib bei Klarzellsarkomen, inflammatorischen myofibroblastären Tumoren (IMFT), alveolären Rhabdomyosarkomen (ARMS) und beim alveolar soft part sarcoma (ASPS) untersucht. In ähnlicher Weise erwies sich in frühen klinischen Studien der Insulin-like-growth-Factor- 1-Rezeptor als ein vielversprechendes Target – etwa bei Ewing-Sarkomen oder Synovialsarkomen.
Ubiquitäre Targets: Targeted therapies von Sarkomen mit komplexen genomischen Aberrationen zielen meist auf ubiquitäre angiogenetische und metabolische Signalkaskaden ab. Inhibitoren der VEGFR- und anderer Tyrosinkinasen wie Sunitinib und Sorafenib sowie der VEGF-Antikörper Bevacizumab haben sich in zumeist kleinen Phase-IIStudien bei Angiosarkomen, ARMS, ASPS, malignen Hämangioperizytomen/solitärem fibrösem Tumor (SFT), zum Teil in Kombination mit Chemotherapien, als ausgezeichnet wirksam erwiesen. Lediglich für einen VEGFRInhibitor, Pazopanib, liegen bislang Daten einer randomisierten Phase-III-Studie (PALETTE) vor: Bei metastasierten, chemotherapievorbehandelten Weichteilsarkomen (nicht alle histologischen Subtypen waren erlaubt!) erwies sich Pazopanib hinsichtlich des progressionsfreien Überlebens (primärer Studienendpunkt) als signifikant wirksamer als Placebo (4,6 vs. 1,5 Monate, p < 0,0001), der Einfluss auf das Gesamtüberleben war in der Zwischenauswertung allerdings (noch) nicht signifikant (11,9 vs. 10,4 Monate, p = 0,1782). Weniger beeindruckend waren die Ergebnisse einer placebokontrollierten randomisierten Phase- III-Studie (SUCCEED) mit dem mTOR-Inhibitor Ridaforolimus als Erhaltungstherapie nach Chemotherapie: Der erzielte, signifikante Benefit hinsichtlich des progressionsfreien Überlebens betrug lediglich knapp über 3 Wochen. Die hervorragenden klinischen Ergebnisse eines anderen mTOR-Inhibitors, Sirolimus, bei malignen perivaskulären Epitheloidzelltumoren, welche typischerweise eine aberrante Aktivierung des mTOR-Pathways durch Verlust des negativen Regulators TSC2 aufweisen, bestätigen jedoch, dass mTOR zumindest bei gewissen Sarkomtypen ein valides Target ist.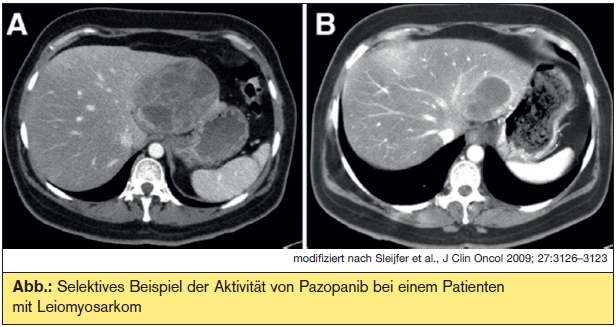
Ausblick
Unser wachsendes Verständnis der einzelnen Sarkomuntertypen zugrunde liegenden häufigen molekularen Veränderungen erlaubt zunehmend den Einsatz zielgerichteter Therapiemodalitä – ten. Zumindest für die genetisch heterogene Gruppe der Sarkome mit komplexen Karyotypen wird wohl die Identifikation von therapeutischen Targets am individuellen Tumor unabdingbar sein.

Ursprünglich erschienen:
UIM 03|2012
UIM 03|2012
Herausgeber: Univ.-Prof. Dr. Günter J. Kreijs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2012-04-13
Zur Ausgabe »
Publikationsdatum: 2012-04-13
Zur Ausgabe »
