


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Spinale Muskelatrophie
Hoffnung durch moderne Therapien
31. Oktober 2025
Die spinale Muskelatrophie (SMA) ist eine seltene, autosomal-rezessiv vererbte Motoneuronerkrankung, die durch Mutationen im SMN1-Gen verursacht wird. Sie führt zu einem fortschreitenden Verlust motorischer Nervenzellen im Vorderhorn des Rückenmarks. Die SMA war bis vor wenigen Jahren nicht kausal behandelbar, durch die Einführung krankheitsmodifizierender Therapien hat sich das Krankheitsbild in den letzten Jahren jedoch grundlegend verändert.
Klassifikation
Die Anzahl der Survival-of-Motor-Neuron-(SMN2-)Genkopien bestimmt den SMA-Typ und damit die Krankheitsausprägung („historische Klassifikation“). Mit etwa 60 % ist der Großteil der Patient:innen von SMA Typ1 betroffen, der (nach Typ 0) die schwerste Form der Erkrankung darstellt. Hier liegt die Lebenserwartung ohne kausale Therapie bei <2 Jahren; bei den weiteren SMA-Typen ist die Lebenserwartung verkürzt bis normal (Tab. 1).
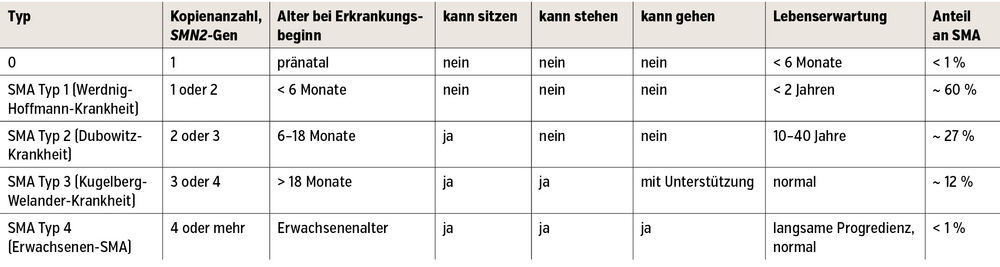
Tab. 1: Überblick über die verschiedenen SMA-Typen (historische Klassifikation)
Die historische Klassifikation berücksichtigt klinische Veränderungen nicht, die mit der Progression der Erkrankung einhergehen. Mit dem Aufkommen der neuen Therapien wurde eine revidierte Klassifikation eingeführt. Diese umfasst die motorische Funktion und unterteilt die Patient:innen in Non-Sitter, Sitter und Walker (Tab. 2).

Tab. 2: Revidierte SMA-Klassifikation
In der klinischen Praxis kommen beide Klassifikationen parallel zur Anwendung.
Erstmanifestationen und Red Flags
In Österreich ist die SMA seit 2021 im Neugeborenen-Screening inkludiert, bisher konnten bereits 49 Patient:innen (Stand September 2025) erkannt und einer Therapie zugeführt werden. Da die Erkrankung erst vor einigen Jahren in das Neugeborenen-Screening aufgenommen wurde, ist davon auszugehen, dass es aktuell nach wie vor nichtdiagnostizierte Patient:innen mit den milderen Verlaufsformen gibt. Hier sind als wichtigste Red Flags für das Vorliegen einer SMA eine proximale Muskelschwäche, der Verlust von motorischen Fähigkeiten sowie Hypo- oder Areflexie zu nennen.
Bei erwachsenen Patient:innen zeigt sich die SMA meist durch symmetrische, vorwiegend proximale Paresen der Extremitätenmuskulatur. Typisch sind Schwierigkeiten beim Treppensteigen oder bei Tätigkeiten über dem Kopf, etwa beim Heben der Arme. Bei schwereren Verlaufsformen entwickelt sich häufig zusätzlich eine axiale Muskelschwäche, die zur Skoliosebildung führen kann. Die Muskeleigenreflexe sind meist abgeschwächt oder erloschen, und es treten Faszikulationen auf, die klinisch sichtbar oder elektromyografisch nachweisbar sind. Beginnt die Erkrankung bereits im Kindesalter, stehen oft Schluckstörungen und eine respiratorische Insuffizienz im Vordergrund, die sich zunächst als nächtliche Hypoventilation mit Hyperkapnie bemerkbar macht.
Differenzialdiagnostische Abgrenzung
Zu den bedeutendsten Differenzialdiagnosen zählen andere neuromuskuläre Erkrankungen wie z. B. die Duchenne- und Becker-Muskeldystrophie (v. a. bei Buben mit proximaler Muskelschwäche). Im Labor findet sich bei der SMA typischerweise eine normale bis gering erhöhte Kreatinkinase (CK), was die Abgrenzung zu Muskeldystrophien ermöglicht. Weitere Differenzialdiagnosen betreffen kongenitale Myopathien und andere Motoneuron-Erkrankungen sowie seltener die myotonische Dystrophie, entzündliche Myopathien, metabolische Ursachen oder (kongenitale) Myasthenien.
Diagnostikpfad
Bei klinischem Verdacht auf das Vorliegen der 5q-assoziierten SMA erfolgt die Sicherung der Diagnose durch eine molekulargenetische Analyse des SMN1-Gens (Deletionsanalyse von Exon 7 ± Exon 8), die Bestimmung der SMN2-Kopienzahl liefert zusätzliche prognostische Informationen. Fällt die Analyse negativ aus, sollte ein erweitertes genetisches Panel durchgeführt werden. In der Elektromyografie (EMG) sind Veränderungen einer andauernden Denervierung (Spontanaktivität durch positive scharfe Wellen, Fibrillationspotenziale und Faszikulationen) sowie chronisch neurogene Veränderungen erkennbar, während die Nervenleitgeschwindigkeit oft unauffällig ist.
Nach gesicherter Diagnose ist eine Basisbefunderhebung zur Verlaufsdokumentation und Therapieplanung sinnvoll. Dazu gehören Lungenfunktionsmessungen, eine Schluckdiagnostik sowie eine orthopädische Beurteilung, insbesondere im Hinblick auf Wirbelsäulen- und Gelenkveränderungen.
Therapie
Die Behandlung der SMA erfordert ein multidisziplinäres Management in spezialisierten Zentren unter Einhaltung der Standards of Care. Diese umfassen u. a. Physiotherapie, Ergotherapie und Logopädie und stellen stets die Basis der Therapie dar. Regelmäßige Verlaufskontrollen am Zentrum sowie eine individuell abgestimmte Behandlung sind für das Management der Erkrankung von großer Bedeutung.
Darüber hinaus sind mittlerweile 3 kausale Therapieoptionen verfügbar, deren gemeinsames Ziel die Herstellung eines voll funktionsfähigen SMN-Proteins ist. Das Antisense-Oligonukleotid Nusinersen ist seit 2017 für alle SMA-Typen und Altersgruppen zugelassen, nach einer Aufsättigungsphase wird der Wirkstoff alle 4 Monate intrathekal verabreicht. Die Gentherapie Onasemnogen-Abeparvovec ist seit 2020 für Patient:innen mit maximal 3 SMN2-Genkopien und bis zu einem Gewicht von 21 kg zugelassen; intakte SMN1-DNA wird über einen viralen Vektor einmalig intravenös verabreicht. Das Small Molecule Risdiplam modifiziert das Spleißen des SMN2-Gens und ist seit 2021 für alle Altersgruppen und Schweregrade zugelassen. Es wird täglich oral als Saft eingenommen; darüber hinaus ist Risdiplam seit Juni 2025 auch in Tablettenform verfügbar.
Nusinersen und Risdiplam wurden in den Zulassungsstudien nur an Kindern getestet, erhielten jedoch für Kinder und Erwachsene eine Zulassung. Nachfolgende klinische Studien belegen eine gute Wirksamkeit beider Substanzen auch bei erwachsenen Patient:innen. Die Therapie sollte jedoch so früh wie möglich erfolgen – im besten Fall bereits präsymptomatisch –, da jede Verzögerung zu fortschreitendem Kraftverlust führt.

Ap. Prof. Priv.-Doz. Dr. Hakan Cetin, PhD
Universitätsklinik für Neurologie, Medizinische Universität Wien

Ursprünglich erschienen:
AEK SH Seltene Erkrankungen|2025
AEK SH Seltene Erkrankungen|2025
KRANKHEITSSTECKBRIEF
- Die SMA zählt mit einer Inzidenz von 1 : 7.000 zu den häufigeren hereditären neuromuskulären Erkrankungen. In Österreich ist die SMA seit 2021 im Neugeborenen-Screening verankert.
- In den allermeisten Fällen kommt es aufgrund einer homozygoten Deletion im SMN1-Gen auf Chromosom 5q13 zu einer Unterproduktion von funktionierendem SMN-Protein, das entscheidend für die Funktion motorischer Nervenzellen ist.
- Verlauf und Prognose hängen von der Anzahl der SMN2-Kopien ab, die den SMA-Typ bestimmen. Vor Einführung der neuen Therapien war die SMA Typ 1 die häufigste genetische Todesursache im Säuglings- und Kleinkindalter, die neuen genbasierten Therapien haben die Prognose wesentlich verbessert.
- Alle kausalen Therapieoptionen führen zur vermehrten Produktion eines funktionsfähigen SMN-Proteins und stabilisieren den Krankheitsverlauf, in vielen Fällen können damit sogar funktionelle Verbesserungen erreicht werden.
Literatur beim Verfasser
Bildnachweis
Vorschaubild: © Julian – stock.adobe.com