


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Bullöse Autoimmundermatosen – Blasenbildung durch Autoantikörper
17. Dezember 2021
Die Therapie besteht in der Kombination von systemischen Glukokortikoiden mit anderen, steroidsparenden Immunsuppressiva. Mit dem Anti-CD20-Antikörper Rituximab lassen sich nunmehr deutlich schnellere und anhaltendere Therapieerfolge erzielen.
Die blasenbildenden Autoimmunerkrankungen können, abhängig von der Lokalisation der Spaltbildung und der Zielantigene, in zwei große Gruppen unterteilt werden: die Pemphigus- und die Pemphigoid-Gruppe.
Pemphigus-Gruppe
Die Pemphigus-Gruppe ist durch Autoantikörper gegen Desmoglein gekennzeichnet, die konsekutiv zu einer akantholytischen Spaltbildung innerhalb der Epidermis führen. Desmogleine sind ein wesentlicher Bestandteil von Desmosomen, die der festen Verankerung der Epidermiszellen untereinander dienen. Klinisch zeigen sich oberflächliche, schlaffe, fragile Blasen oder häufig nur Erosionen. Die Wundheilung erfolgt narbenlos. Großflächige Erosionen, welche im Krankheitsverlauf auftreten, führen oft zu Komplikationen wie Sekundärinfektionen, Wasser- und Proteinverlust. Unbehandelt wiesen Pemphigus-Erkrankungen früher eine hohe Mortalität auf.
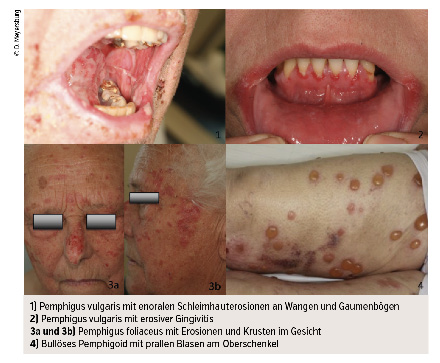
Die wichtigsten Vertreter der Gruppe sind der Pemphigus vulgaris und Pemphigus foliaceus. Die Inzidenz beträgt 0,1–0,5 Erkrankungen pro 100.000 Einwohner. Der Erkrankungsgipfel ist im mittleren bis höheren Lebensalter. Beim P. vulgaris richten sich die Autoantikörper dabei hauptsächlich gegen Desmoglein-3. Klinisch dominieren Erosionen der Mundhöhle und Nasenschleimhäute, die sich daneben auch am Pharynx, Larynx, Ösophagus und genital manifestieren können. In der Hälfte der Fälle, insbesondere wenn auch Autoantikörper gegen Desmoglein-1 hinzukommen („Antigenshift“), zeigen sich zusätzlich schlaffe Blasen und Erosionen an der Haut, die sich großflächig ausbreiten können.
Beim Pemphigus foliaceus, bei welchem primär Autoantikörper gegen Desmoglein-1 nachweisbar sind, treten Erosionen und Krusten hingegen ausschließlich an der freien Haut von meist seborrhoischen Arealen (Gesicht, Kapillitium und Körperstamm) auf.
Im Rahmen von Neoplasien wie Lymphomen, Leukämien oder Thymomen kann es mutmaßlich durch Kreuzreaktion von Tumorantikörpern mit epithelialen Proteinen zu einem paraneoplastischen Pemphigus kommen. Dieser ähnelt klinisch dem Pemphigus vulgaris, wobei die Symptome ungewöhnlich schwer verlaufen und therapieresistent sind.
Pemphigoid-Gruppe
Die Erkrankungen der Pemphigoid-Gruppe weisen hingegen eine subepidermale und somit tiefere Spaltbildung innerhalb der Basalmembran auf. Die Autoantikörper richten sich dabei gegen die Hauptantigene an den Hemidesmosomen BP230 und BP180. Klinisch kommt es aufgrund der histologisch tieferen Spaltbildung zum Auftreten von prall gespannten Blasen. Der Hauptvertreter der Pemphigoid-Gruppe ist das bullöse Pemphigoid, welches mit einer altersabhängig rasch steigenden Inzidenz von 10–40 pro 100.000 Einwohner auch die häufigste bullöse Autoimmundermatose des Erwachsenenalters ist. Der Erkrankungsgipfel liegt zwischen dem 70. und 80. Lebensjahr. Die Blasen weisen meist einen serösen oder hämorrhagischen Inhalt auf und treten bevorzugt in den großen Falten, am Abdomen und an Armen und Oberschenkeln auf. Meist erfolgt die Wundheilung ebenfalls narbenlos. Ein nichtbullöses Prodromalstadium mit papulovesikulösen oder urtikariellen Plaques kann manchmal auftreten. Eine Assoziation mit Medikamenten (insbesondere Antidiabetika der Gruppe DPP-4-Inhibitoren und antitumorale Immuntherapie mit PD-1-Inhibitoren) ist möglich.
Diagnostik
Die Diagnostik der bullösen Autoimmundermatosen erfolgt mittels Histologie sowie direkter und indirekter Immunfluoreszenzuntersuchung. Die Histologie dient hauptsächlich zur Unterscheidung der Ebene der Spaltbildung (intra- vs. subepidermal bzw. Pemphigus vs. Pemphigoid). Mit der direkten Immunfluoreszenz als spezifischste Goldstandard-Methode lässt sich bei der Pemphigus-Gruppe eine netzförmige interzelluläre Immunglobulinablagerung bzw. bei der Pemphigoid-Gruppe eine lineare IgG-/IgA- und Komplementablagerung entlang der Basalmembran nachweisen. Die serologische Diagnostik zum Nachweis der unterschiedlichen Autoantikörper kann mittels indirekter Immunfluoreszenz (auf humaner Spalthaut oder Affenösophagus) oder spezifischen ELISA erfolgen. Seltenere Zielantigene, für welche kein ELISA verfügbar ist, können mittels Immunoblot oder auf kultivierten Keratinozytenextrakten detektiert werden.
Therapie
Die Therapie der Wahl bei den bullösen Autoimmundermatosen ist initial die systemische Gabe von Glukokortikoiden, wobei die Dosierung bei der Pemphigus-Gruppe aufgrund des schwereren Verlaufes deutlich höher ist (1–2 mg/kg/d Prednisolon) als bei den Pemphigoiden (0,5–1 mg/kg/d Prednisolon). Diese werden in der Regel mit oralen Immunsuppressiva (Azathioprin, Mykophenole) oder Immunmodulatoren (Doxycyclin, Dapson) kombiniert. Um das Nebenwirkungsspektrum langfristig hochdosierter, systemischer Steroidtherapie insbesondere bei Risikogruppen zu vermeiden, empfiehlt die Leitlinie für das milde bullöse Pemphigoid lediglich die Ganzkörperanwendung von topischem Clobetasolpropionat 0,05 % in ausschleichender Menge und Anwendungsfrequenz. Seit 2019 ist von der europäischen Arzneimittelbehörde der Anti-CD20-Antikörper Rituximab für den moderaten und schweren Pemphigus vulgaris zugelassen, was die therapeutische Versorgung der Patienten deutlich verbessert hat. Diese zielgerichtete Therapie hat sich als signifikant effektiver und sicherer erwiesen als langjährige Therapien mit systemischem Prednisolon.
Wissenswertes für die Praxis
- Bullöse Autoimmundermatosen sind Erkrankungen mit Autoantikörpern gegen Adhäsionsproteine der Haut und Schleimhäute.
- Die wichtigsten und häufigsten Vertreter sind der Pemphigus vulgaris und das bullöse Pemphigoid.
- Zur Diagnostik benötigt man eine Histologie, eine periläsionale direkte Immunfluoreszenz und einen serologischen Autoantikörpernachweis.
- Die Therapie besteht in der Kombination von systemischen Glukokortikoiden mit anderen, steroidsparenden Immunsuppressiva. Mit dem Anti-CD20-Antikörper Rituximab lassen sich nunmehr deutlich schnellere und anhaltendere Therapieerfolge erzielen.

AutorIn: OA Dr. Damian Meyersburg
Universitätsklinik für Dermatologie und Allergologie, Universitätsklinikum Salzburg, Salzburger Landeskliniken
© Hametner Rudolf

AutorIn: Dr. Laura Krecu
Universitätsklinik für Dermatologie und Allergologie, Universitätsklinikum Salzburg, Salzburger Landeskliniken
© privat

Ursprünglich erschienen:
AEK 24|2021
AEK 24|2021