


























































































Die österreichische Fachzeitschrift für Rheumatologie mit wissenschaftlichen Updates zu Pathogenese, Diagnostik und Therapie sowie DFP-Fortbildung in jeder Ausgabe.
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Unter dem Überbegriff „Vaskulitis“ wird eine sehr heterogene Gruppe von entzündlichen Erkrankungen zusammengefasst, wobei grundsätzlich Gefäße aller Art und Größe befallen sein können und auch das Beschwerdebild sehr unterschiedlich ausfallen kann.
Somit kann auch die Nierenbeteiligung sehr unterschiedlich sein: Beim Befall großer Gefäße wird der Blutzufluss behindert und es kann zum Niereninfarkt oder zur ischämischen Schädigung kommen. Sind die glomerulären Kapillaren betroffen, führt dies zu einer nekrotisierenden Glomerulonephritis.
Besonders dieser zuletzt genannte Zustand ist typisch für eine Gruppe der Vaskulitiden, die mit einer systemischen nekrotisierenden Vaskulitis der kleinen Gefäße einhergehen und Organmanifestationen in verschiedenen Organen wie der Lunge und eben der Niere hervorrufen können.
Diese Systemvaskulitiden sind häufig von einer Erhöhung der antineutrophilen zytoplasmatischen Antikörper (ANCA) begleitet und werden als ANCA-assoziierte Vaskulitiden (AAV) bezeichnet. Gemeinsam ist ihnen auch die sehr ähnliche klinische und histologische Nierenbeteiligung (s. u.). Die Art und Häufigkeit der Lungenbeteiligung und andere klinische Kennzeichen unterscheiden allerdings den M. Wegener (WG), die mikroskopische Polyangiitis (MPA) und das Churg-Strauss-Syndrom (CSS). Nach den Chapel-Hill-Consensus-Kriterien von 1994 gehören die AAV zu den primären Vaskulitiden, die kleine Gefäße und mittelgroße Arterien befallen1, 2.
Die Häufigkeit der Nierenbeteiligung bei den AAV schwankt von fast 100 % bei MPA und 70 % bei M. Wegner zu etwa 25 % beim Churg-Strauss-Syndrom.
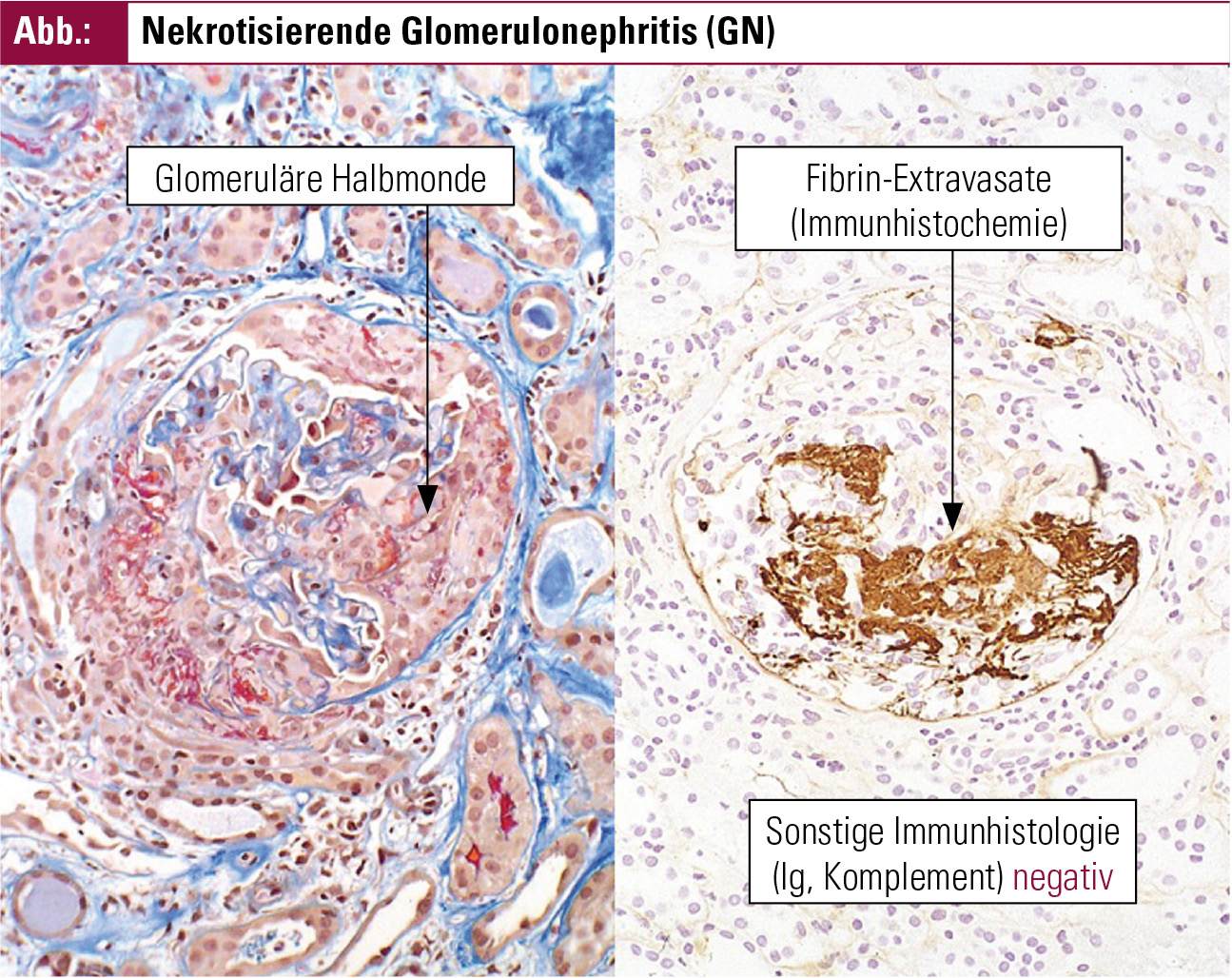
Die wichtigste klinische Präsentation der Nierenbeteiligung zeigt sich in Form einer rapid-progressiven Glomerulonephritis (RPGN). Dabei kommt es zu einer raschen Abnahme der glomerulären Filtrationsrate um zumindest 50 % und zur Bildung von glomerulären Halbmonden (s. u.). Im Harnsediment bzw. 24h-Harn zeigt sich eine mikroskopische Hämaturie (häufig mit Erythrozytenzylindern) und eine Proteinurie, meist unterhalb von 3 g/24 h.
Die RPGN ist ein internistischer Notfall, 60 % der Patienten werden dialysepflichtig.
Der Nachweis einer Nierenbeteiligung im Sinne einer nekrotisierenden Glomerulonephritis erfolgt bei klinischem Verdacht durch eine Nierenbiopsie. Das histologische Kennzeichen der GN bei AAV besteht aus Nekrosen der glomerulären Schlingen, die in der Regel nicht jeder Glomerulus betreffen (d. h.„fokal“ sind) und nur Teile eines Glomerulus betreffen („segmental“); sie können mit oder ohne Halbmondbildung auftreten.
Unter Halbmondbildung versteht man die im Glomerulus stattfindende intrakapilläre und extrakapilläre Proliferation von mononukleären Zellen, die sich schließlich halbmondförmig um das Schlingenkonvolut anordnen und zur Fibrose (zunächst) innerhalb der Bowmann’schen Kapsel führen. Daneben können völlig unauffällige Glomeruli existieren. Sind mehr als 50 % der Glomeruli von einer Halbmondbildung betroffen, entspricht der klinische Verlauf der Nierenfunktion meist einer rapid-progressiven GN (RPGN, s. o.).
In der Immunhistochemie finden sich kaum Immunkomplex- und Komplementfaktor-Ablagerungen, sie ist „pauci-immun“ (zitiert nach de Groot K, 2006)2, siehe Abbildung.
Selten findet sich eine obstruktive Uropathie wegen einer ureteralen granulomatösen Vaskulitis bei SCC oder GPA, selten gibt es eine isolierte interstitielle Nephritis, oder – bei Beteiligung größerer Gefäße – eine Ruptur eines arteriellen Aneurysmas1.

Zum Thema der Therapie sind in den letzten Jahren nicht zuletzt aufgrund der besseren Klassifikation der Erkrankungen eine Reihe von hochkarätigen Publikationen erschienen, welche die therapeutischen Möglichkeiten erweitert und abgesichert haben. Die Behandlung der GN bei AAV teilt sich in die Therapie der akuten GN (Induktionstherapie) und in eine Erhaltungstherapie, wenn die akute Phase abgeklungen ist und Remission erzielt werden konnte.
Zur Induktionstherapie werden Glukokortikoide und Cyclophosphamid (CYC; Endoxan®) verabreicht, bis nach meist 3–6 Monaten eine Remission erzielt werden kann3. Dabei ist orales CYC genauso wirksam wie intravenöse CYC-Pulstherapie, wobei Letztere eine geringere kummulative CYC-Dosis zur Folge hat und daher vorzuziehen ist4, 5.
Patienten mit einem Serumkreatinin von > 500 µmol/l (5,7 mg/dl) sollen nach den Empfehlungen der European Vasculitis Study Group (EUVAS) zusätzlich auch mittels Plasmaaustausch behandelt werden6, 7.
In rezenten Studien hat sich als Alternative zur CYC-Therapie auch die Wirksamkeit von Rituximab (Anti-CD20-Ak) gezeigt, das vor allem bei der Behandlung von jungen Patienten (und hier speziell bei jungen Patientinnen zur Erhaltung der Fertilität) bevorzugt werden sollte8–10. Auch CYC-refraktäre Patienten haben von Rituximab profitiert8, 11, 12.
Erhaltungstherapie
Azathioprin (AZA, Imurek®) ist so gut wie orales CYC in der Remissionserhaltung (aber weniger toxisch) und effektiver als Mykophenolat-Mofetil (MMF, Cellcept®)3 ,13; Methotrexat (MTX) ist in der Erhaltungstherapie der AAV grundsätzlich gleich wirksam wie AZA, darf aber wegen der Gefahr der Akkumulierung bei eingeschränkter Nierenfunktion nicht angewandt werden; auch bei Patienten in renaler Remission sollte AZA bevorzugt werden14.
Auch Rituximab hat gute Ergebnisse in der Remissionserhaltung in nicht kontrollierten Studien gezeigt8, 11.
Die ideale Dauer der Erhaltungstherapie ist Gegenstand von Debatten, sie sollte zumindest 1 Jahr betragen1, jedoch erscheint eine Fortführung oft sinnvoll und muss letztlich individuell entschieden werden.
Begleitende Therapie und Kontrollen
Wichtig sind natürlich die Prophylaxe und das Monitoring bzgl. Infektionen (Pneumocystis jiroveci, opportunistische und Pilzinfektionen).
Bei Proteinurie (bzw. arterieller Hypertonie) sollte man ACE-Inhibitoren oder AT-II-Blocker geben. Engmaschige Kontrollen der Nierenfunktionsparameter inklusive Harnsediment und Proteinurie bzw. des Blutbilds unter immunmodulatorischer Therapie sind geboten.


