


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Biosimilars – Nicht variabler als das Original
26. Dezember 2012
Der medizinische Fortschritt durch Biologika steht außer Zweifel. Sie sind ein ganz wesentlicher Schritt auf dem Weg zu einer individualisierten Medizin, stellen die Gesundheitssysteme aber in puncto Kosten vor Herausforderungen. Nach Ablauf des Patentschutzes ermöglichen die Nachfolgeprodukte originärer Biologika den Einsatz dieser Substanzen mit geringerem Kostenaufwand. Dadurch kann der Nutzen der biologischen Wirkstoffe nicht nur einem weiteren Personenkreis zugänglich gemacht werden, sondern es wird auch Budget für den Einsatz von Innovationen frei. Biosimilars sind so indirekt Treiber biologischer Innovationen.
Nicht jeder Biologikumnachfolger ist ein Biosimilar: Als erstes Biosimilar in der EU wurde Somatropin von Sandoz im Jahr 2006 zugelassen. Im Jahr 2007 folgte das erste Erythropoetin-Biosimilar, ebenfalls von Sandoz, und im Jahr 2008 wurde das erste Filgrastim-Biosimilar zugelassen. Die Verfügbarkeit von Filgrastim-Biosimilars führte in Teilen Europas zu einer deutlichen Zunahme des Einsatzes von granulozytenkoloniestimulierenden Faktoren.
Die Herstellung dieser Nachfolgersubstanzen ist weit aufwändiger als die eines einfachen chemischen Arzneimittels. Denn Biologika sind sehr komplexe Substanzen, deren Eigenschaften in hohem Maß durch den Herstellungsprozess bestimmt werden. Nachfolgesubstanzen sind keine identischen Kopien des Originals, sondern „similar“, also ähnlich.
In Europa werden diese Nachfolgeprodukte nur dann als „Biosimilars“ zugelassen, wenn sie die strengen Vorgaben der EMA erfüllen.
„Biosimilar“ ist also kein Synonym für jede Nachfolgesubstanz eines Biologikums, sondern ausschließlich eine Bezeichnung für hochwertige Biologikanachfolger mit EMA-Zulassung.
Die EMA hat auf dem Gebiet der Zulassung von Biosimilars weltweit eine Vorreiterrolle. Die rechtliche Grundlage, die überarbeitete Direktive 201/83/EC, umfasst zahlreiche generelle und produktspezifische Guidelines und verfolgt den Zugang der vollständigen Evidenz. Eine Extrapolation auf andere zugelassene Indikationen ist möglich. Von der EMA werden nur qualitativ hochwertige Nachfolgepräparate von Biologika zugelassen, und es wurden bereits zahlreiche eingereichte Substanzen abgelehnt, die die strengen Kriterien der EMA für den Nachweis einer Gleichwertigkeit mit der Originalsubstanz nicht erfüllten. Derzeit werden 7 neue potenzielle Biosimilars von der EMA evaluiert.
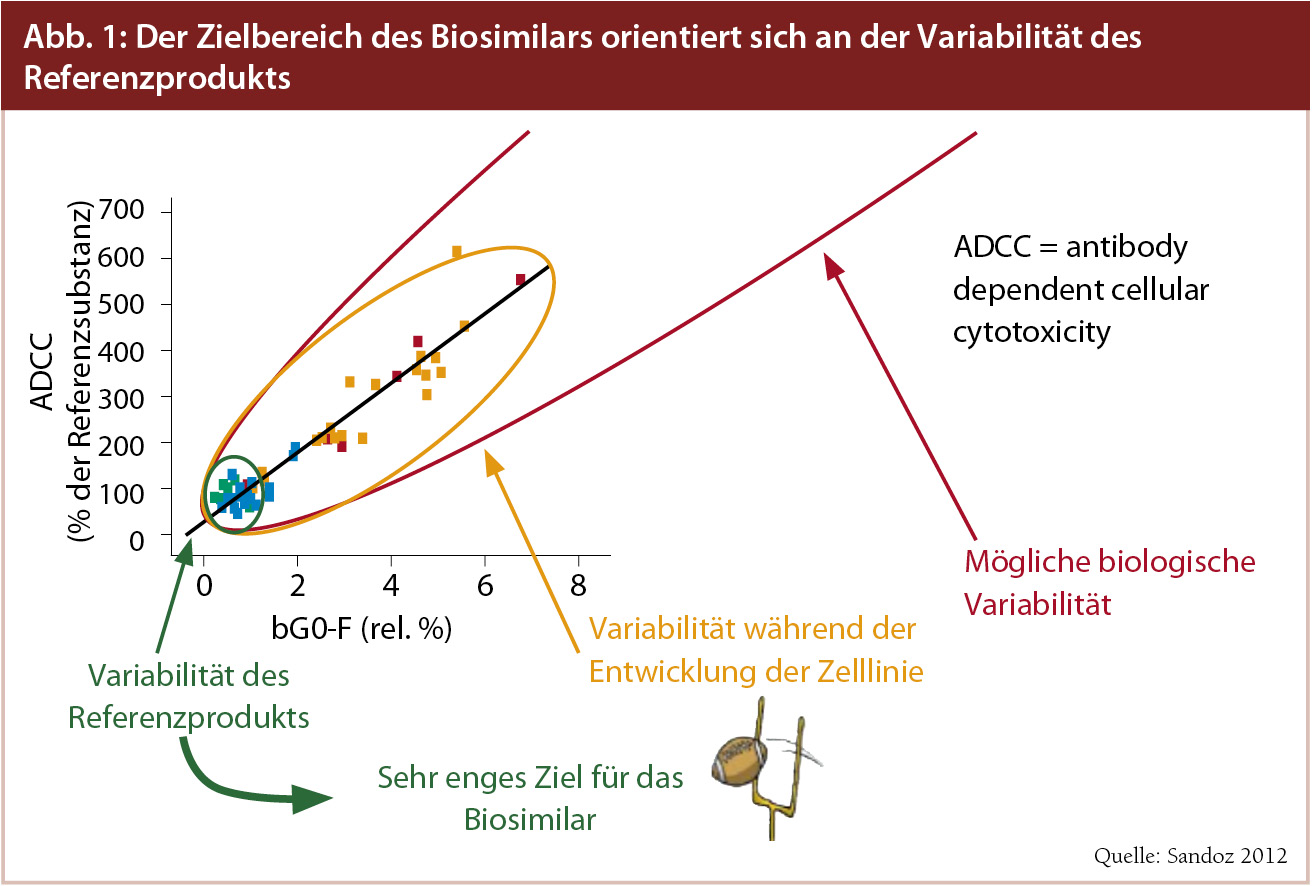
Variabilität als generelles Merkmal biologischer Arzneien: Mit hochspeziellen Analysemethoden können Biologika heute sowohl strukturell als auch funktionell genauestens charakterisiert werden, betont Joerg Windisch, PhD., Chief Science Officer von Sandoz, dem weltweit führenden Biosimilarhersteller. Dadurch kann die Forderung der EMA für die Zulassung eines Biosimilars erfüllt werden: der Nachweis der Äquivalenz des biologischen Nachfolgepräparats mit der Originatorsubstanz hinsichtlich Reinheit, Sicherheit und Wirksamkeit. Hier kommt eine Eigenschaft von biologischen Arzneimitteln ins Spiel, die diese von einfachen chemischen Substanzen unterscheidet: die Variabilität. Zielbereich für das Biosimilar ist die Variabilität der Originatorsubstanz. Und diese ist, wie Abbildung 1 zeigt, sehr klein. Dass auch die Eigenschaften der Originatorsubstanzen gewissen Schwankungen unterworfen sind, zeigten Schiestl et al.1 in ihrer in Nature Biotechnology veröffentlichten Arbeit am Beispiel von Darbepoetin alfa (Aranesp®), Etanercept (Enbrel®) und Rituximab (MabThera®). Wie Windisch erläutert, ist es heute mit hochspezifischen Analysemethoden möglich, verschiedene Chargen ein und desselben Biologikums zu unterscheiden und selbst geringfügige Änderungen im Herstellungsprozess zu erkennen. Auch Originatoren müssen für das Fortbestehen der Zulassung nach Änderungen des Produktionsprozesses die Vergleichbarkeit mit dem ursprünglich zugelassenen Biologikum mit den gleichen Instrumenten und Verfahren nachweisen, die im Rahmen der Zulassung von Biosimilars angewendet werden.
Übereinstimmender „Fingerabdruck“ gefordert: Für die Zulassung eines Biosimilars muss nicht nur gezeigt werden, dass die Proteinsequenz von Nachfolgeprodukt und Originatorsubstanz identisch ist, auch die Strukturen höherer Ordnung und die Funktionalität müssen vollständig übereinstimmen. Die Bestimmung der Eigenschaften des herzustellenden similaren Biologikums beginnen mit dem Screening geeigneter Wirtszellen, des Transfektionspools und in weiterer Folge eines geeigneten Klons, wobei die Variabilität des gewünschten Produkts in genannter Reihenfolge abnimmt. Der Aufwand für die Auswahl des letztlich verwendeten Zellklons ist extrem hoch. Auch die Entwicklung des Kulturmediums und des Bioprozesses und ganz zuletzt der Aufreinigungsprozesse hat einen, wenn auch deutlich geringeren Einfluss auf die Vergleichbarkeit. Abbildung 2 veranschaulicht, wie weit die Abklärung der Übereinstimmung von Originatorsubstanz und Biosimilar, aber auch einer Originatorsubstanz vor und nach einer Änderung im Herstellungsprozess, heute geht. Es werden quasi die „Fingerabdrücke“ der beiden Substanzen miteinander verglichen. Die Schlüsse, die aus diesen Analysen gezogen werden können, gehen sogar weit über die Erkenntnisse hinaus, die klinische Head-to-Head-Studien liefern, betonte Windisch. Anhand klinischer Studien gelingt es oft nicht, einen Unterschied im klinischen Effekt von Substanzen zu zeigen, während beispielsweise anhand eines SPR-Sensorgramms sehr genau zwischen Biologika differenziert werden kann.
Das von der EMA vorgeschriebene Ausmaß der klinischen Prüfung eines zur Zulassung eingereichten Biologikanachfolgeprodukts ist abhängig von den Erkenntnissen aus der physiochemischen und biologischen Charakterisierung, der präklinischen Daten und der Pharmakokinetik und -dynamik. Die klinischen Studien bestätigen die Vergleichbarkeit nur. Ein De-novo-Beweis der Wirksamkeit ist nicht notwendig.
Umfangreiche Expertise aus Österreich: Mit Sandoz besitzt ein in Österreich tätiges Unternehmen umfangreiche Expertise auf dem Gebiet der Biologika. Seit den 1980er-Jahren werden in Kundl in Tirol moderne Biologika entwickelt – von Peptiden bis zu monoklonalen Antikörpern. Das Unternehmen verfügt als Pionier auf diesem Gebiet über umfangreiche Expertise und hat als weltweit einziges Unternehmen 3 zugelassene Biosimilars in seinem Portfolio. Kundl ist nicht nur das Biopharma-Kompetenzzentrum von Novartis, hier erfolgt auch eine umfangreiche Lohnfertigung für Hersteller von Originalprodukten. Gleichzeitig wird intensiv an der Entwicklung von zahlreichen weiteren Biosimilars geforscht, wobei der Fokus auf monoklonalen Antikörpern liegt. Die Forschungsprogramme zu einem Rituximab-Biosimilar und einem Pegfilgrastim-Biosimilar sind bereits weit fortgeschritten.
Quelle: Sandoz Biosimilars Media Day, Kundl, 20. 11. 2012
1 Schiestl M et al., Nature Biotechnology 2011; 29:310–312

Ursprünglich erschienen:
GA 06|2012
GA 06|2012