


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
„Evolving Landscapes“ bei Weichteilsarkomen
24. Februar 2012
Das therapeutische Umfeld, an dem sich neue Substanzen messen, besteht nach wie vor in der First-Line-Chemotherapie aus Doxorubicin oder Ifosfamid entweder als Monotherapie oder in Kombination, d. h. aus zwei Referenzsubstanzen, die in den 1970er und 80er Jahren etabliert wurden und als Monotherapie Ansprechraten von 10–25 % erreichen (Verweij; The Oncologist 2008). Im Jahr 2009 wurde Trabectedin (Yondelis®) als neues Zytostatikum zugelassen, das in einer Phase-II-Studie bei 270 Patienten mit Liposarkomen und Leiomyosarkomen als Second-Line-Therapie untersucht wurde und in dieser Studie das progressionsfreie Überleben verlängern konnte (Demetri et al., J Clin Oncol 2009). Verabreicht als 24-Stunden-Infusion alle drei Wochen betrug der Anteil progressionsfreier Patienten (PFR) nach 3 Monaten 51,5 % und nach 6 Monaten 35,5 %, womit die EORTC-Screening- Kriterien für eine aktive Substanz im Einklang mit drei weiteren vorangehenden Phase-II-Studien erfüllt waren.
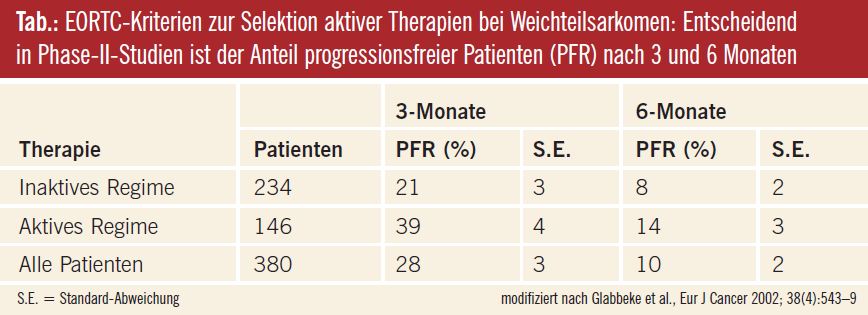
PFR nach EORTC: Im Jahr 2002 wurde von der EORTC das progressionsfreie Intervall zu einem bestimmten Zeitpunkt, 3 und 6 Monate nach Therapiebeginn, als primärer Endpunkt für Phase-II-Studien vorgeschlagen, was insbesondere für nichtzytotoxische Substanzen wesentlich ist, deren Wirkmechanismus nicht unbedingt zu einer fassbaren Tumorverkleinerung führt (van Glabbeke et al., Eur J Cancer 2002). Bleibt ein bestimmter Anteil der Patienten 3 Monate nach Therapiebeginn progressionsfrei (progression free survival rate, PFR), wären weitere Untersuchungen in randomisierten Phase-II-Studien gerechtfertigt. (vgl. Tab. und Kommentar OA Dr. Ploner).
Histologie, molekulare Tumorbiologie: Die Frage, ob man so seltene Tumoren immer weiter aufsplittert („Splitters“) oder zur größtmöglichen Gruppe zusammenfasst („Lumpers“), beantwortet sich durch eine nichtreproduzierbare therapeutische Wirkung, wenn Besonderheiten einzelner Subspezialitäten nicht ausreichend berücksichtigt werden. Interessanterweise ist es der Effektivität von Imatinib bei gastrointestinalen Stromatumoren zu verdanken, dass diese Form von Weichteiltumoren, die früher zur Gruppe der Leiomyosarkome gerechnet wurde, heute als eigenständige Tumorentität geführt wird und klinische Studien speziell darauf zugeschnitten sind (Erstbeschreibung von Kit-Mutationen durch Hirota im Jahr 1998, erster therapeutischer Einsatz von Imatinib bei GIST-Patienten im Jahr 2000, Zulassung im Jahr 2002). Konsequenterweise war Imatinib in einer Phase-II-Studie bei 190 Patienten mit 10 anderen Sarkom-Subtypen (z. B. Ewing-Sarkome, Angiosarkome, Fibrosarkome, Synovialsarkome) nicht in dem Maß effektiv oder ineffektiv (Chugh et al., J Clin Oncol 2009), was die Besonderheit von Kit-Mutationen als treibende Kraft bei GIST unterstreichen kann (oncogene addiction). Chugh et al. weisen z. B. auch darauf hin, dass Doxo rubicin, die Standard-First-Line-Therapie von Weichteilsarkomen, bei GIST keine Aktivität zeigt.
Angiogeneseabhängigkeit: Weichteilsarkome sind stark vaskularisierte Tumoren, wobei VEGF, PDGF oder die Mikrogefäßdichte in verschiedenen Studien mit der Tumorhistologie und der Prognose assoziiert wurden (z. B. Chao et al., Ann Surg Oncol 2001; Yudoh et al., Br J Cancer 2004). Der Einsatz von Angiogenesehemmern wie Bevacizumab, Cediranib, Pazopanib, Sorafenib oder Sunitinib war damit naheliegend und hat entsprechend der biologischen Variabilität zu unterschiedlichen Beobachtungen in Phase-II-Studien geführt: So zeigte sich exemplarisch eine Aktivität mit Sorafenib insbesondere bei Angiosarkomen, die sich inhärent als Target für verschiedene Angiogenesehemmer anbieten (Maki et al., J Clin Oncol 2009). Eine Aktivität von Sunitinib wurde insbesondere bei solitär fibrösen Tumoren hervorgehoben, die PDGFR und VEGFR exprimieren (Hämangioperizytom); darüber hinaus bei desmoplastischen kleinen, rundzelligen Tumoren, die durch ein Fusionsonkogen (EWS-WT1) charakterisiert sind, das wieder zur Aktivierung von PDGF-Rezeptoren führt (George et al, J Clin Oncol 2009). Schließlich ist am ASCO 2011 eine Studie mit Cediranib, einem VEGFR- 1,-2,-3-Hemmer, beim alveolären Weichteilsarkom (alveolar soft part sarkoma, ASPS) präsentiert worden. In dieser umschriebenen Indikation, von der in den USA jährlich etwa 100–150 Fälle zu erwarten sind (< 1 % aller Weichteilsarkome), hat Cediranib einen hohen Anteil partieller Remissionen erreichen können – wobei Sunitinib hier ebenfalls besonders aktiv scheint. Diese Beispiele, die noch durch Studien mit mTOR-Inhibitoren wie Ridaforolimus (Phase-III-Studie SUCCEED) erweitert werden können, zeigen jedenfalls, dass Weichteilsarkome im Rahmen der erfolgten Subtypisierung in einem zunehmend kompetitiven Umfeld nunmehr auch therapeutisch erschlossen werden – wenngleich der Aufwand hoch ist und die an sich rare Entität zu einem Dschungel an Entitäten wurde („Targeting molecular pathways in the jungle of soft tissue sarcoma histology“, ESMO-Symposium 2010).
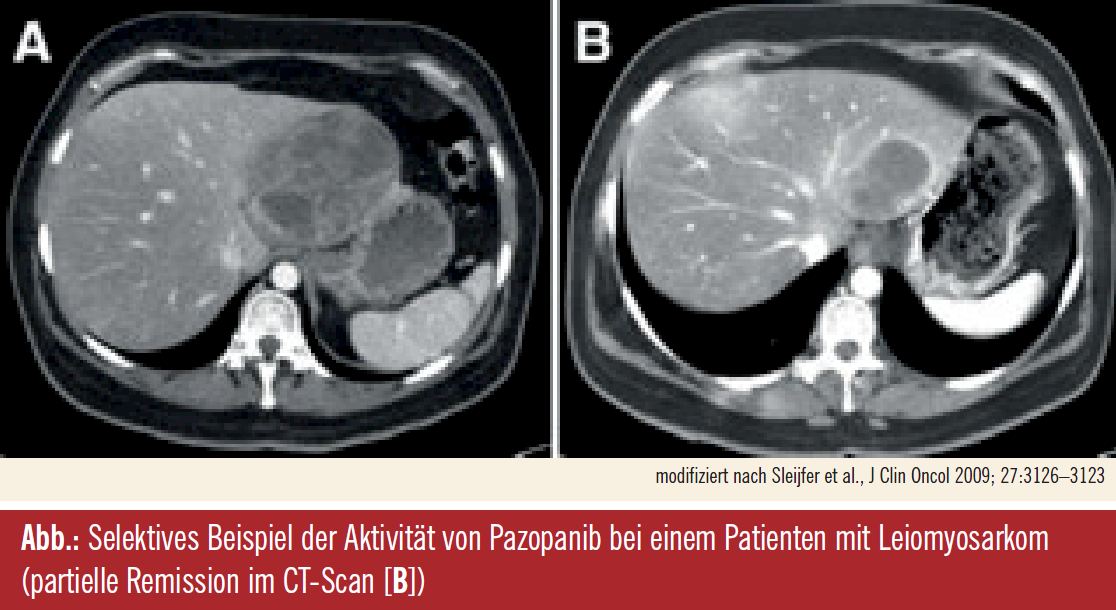
PALETTE, Zulassungsstudie mit Pazopanib1: In diesem Kontext am weitesten fortgeschritten ist die Entwicklung von Pazopanib, einem Multikinasehemmer gegen VEGFR, PDGFR und c-Kit, von dem – nach positiven Ergebnissen einer placebokontrollierten Phase-III-Studie (PALETTE) – die Zulassung in der zweiten Hälfte dieses Jahres erwartet wird. Die Zulassungsstudie folgte einem EORTC-Screening-Programm (Studie 62043), in dem Pazopanib bei Patienten mit fortgeschrittenen Leiomyosarkomen, Synovialsarkomen und anderen Subtypen mit Ausnahme von Liposarkomen nach den EORTC-Kriterien effektiv war (Sleijfer et al., J Clin Oncol 2009). Der Multikinasehemmer wurde bei chemotherapieprogredienten Patienten in einer Dosis von 800 mg täglich bis zur erneuten Progression oder dem Auftreten inakzeptabler Nebenwirkungen verabreicht; primärer Studienendpunkt war das progressionsfreie Überleben, sekundärer Endpunkt das Gesamtüberleben. Insgesamt wurden 369 Patienten mit 8 verschiedenen Sarkomsubtypen rekrutiert und nach dem Performancestatus und der Anzahl ihrer Vortherapien stratifiziert. Die am ASCO 2011 präsentierten Ergebnisse erstreckten sich auf ein medianes Follow-up von 15 Monaten, in dem das progressionsfreie Überleben mit Pazopanib von 1,5 auf 4,6 Monate verlängert wurde, entsprechend einer Reduktion des Progressionsrisikos um 69 % (HR 0,31; p < 0,0001), wobei die Aktivität in allen histologischen Strata evident war. Aus der Interimsanalyse geht eine Reduktion des Mortalitätsrisikos um 17 % (p = 0,17) hervor, die im Zusammenhang mit einer Reihe an Postprotokolltherapien (z. B. Trabectedin, Gemcitabin, Docetaxel, Anthrazyklin) interpretiert wurde; bei knapp der Hälfte der Patienten wurde eine nachfolgende Therapie verabreicht, möglich waren bis zu vier. Die Mehrheit der Patienten (67 %) profitierte von einer Krankheitsstabilisierung, bei einem Teil der Patienten (6 %) wurden partielle Remissionen erreicht – wobei Tumorstabilisierungen letztlich den Therapieerfolg neuer Substanzen ausmachen, immer unter Berücksichtigung, dass die therapeutisch induzierte Tumorstabilisierung länger dauern sollte als man von der Biologie des Tumors erwarten kann. Grad-1/2-Tumoren (in dieser Studie etwa bei einem Drittel der Patienten), sind oft langsam progredient und können über längere Zeit indolent verlaufen. Die Therapiedauer war in der Pazopanib-Gruppe doppelt so lange als im Placebo-Arm und die Dosisintensität war hoch (96,3 %), auch wenn im Lauf der Behandlung Therapieunterbrechungen oder Dosisreduktionen erforderlich waren. Die häufigsten Grad-3/4-Nebenwirkungen waren Fatigue, Diarrhö, Hypertonie und Anorexie, wobei Fatigue als Nebenwirkung einer Substanz, deren Benefit aus der Langzeitanwendung resultiert und die im besten Fall dauerhaft eingenommen wird, à la longue beachtet werden soll. Kardiale Nebenwirkungen wurden als seltenes Ereignis beschrieben und verliefen zumeist asymptomatisch. Als Schlussfolgerung wurde festgehalten, dass Pazopanib bei Anthrazyklin-vorbehandelten Patienten mit metastasierten Weichteilsarkomen eine aktive Substanz ist und sich als zielgerichtete neue Therapie in dieser Indikation empfiehlt.
1 PALETTE: A randomized, double-blind, phase III trial of pazopanib versus placebo in patients with soft-tissue sarcoma whose disease has progressed during or following prior chemotherapy – An EORTC STBSG Global Network Study (EORTC 62072). Van Der Graaf WT et al., J Clin Oncol 2011; 29 (suppl; abstr LBA10002).

Ursprünglich erschienen:
SO 01|2012
SO 01|2012
Herausgeber: Univ.-Prof. Dr. Christoph Zielinksi, Univ.-Prof. Dr. Markus Raderer
Publikationsdatum: 2012-02-24
Zur Ausgabe »
Publikationsdatum: 2012-02-24
Zur Ausgabe »